
非洲猪瘟病毒拮抗宿主抗病毒免疫分子机制研究进展
2022-12-09 10:07:16时梦晗胡永新张友明吴晓东曹晶晶
中国动物检疫 2022年12期
时梦晗,胡永新,张友明,吴晓东,曹晶晶
(1.山东大学微生物技术研究院,微生物技术国家重点实验室,山东青岛 266237;2.中国动物卫生与流行病学中心,山东青岛 266032)
非洲猪瘟(African swine fever,ASF)是由非洲猪瘟病毒(African swine fever virus,ASFV)引起的一种高度接触性传染病,急性ASF 致死率可高达100%。1921 年ASF 首次被发现于肯尼亚,到目前为止,已有68 个国家发生该病[1]。其中,我国作为世界上最大的猪肉生产国和消费国受其影响尤为严重[2]。充分了解ASFV 的致病机制有助于设计有效的疫苗进行疾病防控[3]。
ASFV 是一种核质巨DNA 病毒,也是目前唯一的虫媒DNA 病毒[4],其宿主包括家猪、野猪和软蜱[5]。ASFV 颗粒是一个多层的二十面体结构,直径为260~300 nm,病毒基因组为双股线状DNA,其中央保守区长约125 kb,两端为可变区[6]。ASFV 基因组编码的蛋白复杂多样,目前已知的有150 多种编码蛋白且部分蛋白的功能已被鉴定,这些蛋白参与病毒粒子结构组成、基因组转录复制和翻译、病毒入侵和细胞免疫功能调控等[7]。由此可见ASFV 基因组十分庞大且编码蛋白复杂多样。ASFV 主要感染单核-巨噬细胞系统,通过网格蛋白介导的内吞作用或巨胞饮作用完成入侵过程[8],进一步完成复制、组装和子代病毒颗粒的释放。
在感染过程中,为抵御病毒攻击,细胞的免疫系统被激活,同时,为促进自身的存活和复制,病毒进化出逃避或抑制宿主免疫反应的机制[9]。ASFV 编码多种免疫逃逸蛋白以拮抗宿主免疫反应,按参与的细胞反应事件可分为4 类,包括调控宿主细胞蛋白表达系统及细胞因子转录的蛋白(pA238L)、抑制I 型干扰素(IFN-I)信号通路的蛋白(pMGF360,pMGF505,pI329L)、调控细胞程序性死亡的蛋白(p54、pA179L、pA224L和pEP153R)以及其他免疫抑制蛋白(CD2v、pL83L)[4],以上蛋白拮抗宿主抗病毒免疫反应事件既相互独立又协同合作,共同构建拮抗宿主免疫反应的分子网络。本文对ASFV 的结构和生命周期进行简要介绍,并对病毒编码蛋白逃避宿主免疫应答反应的分子机制进行详细归纳,为未来ASFV致病机制研究和疫苗研发提供参考。
1 ASFV 基因组和粒子结构
ASFV 粒子呈二十面体,基因组长为170~190 kb,包含150~167 个紧密排列的开放阅读框,基因组包括中央保守区域和两侧的5 个多基因家族(multigene families,MGF),多基因家族的缺失或重复则导致了不同分离株基因组长度不同[10]。ASFV 呈多层结构,从外到内依次是外包膜、衣壳、内膜、核壳和类核[4,11]。
病毒粒子的最外层为外包膜,是病毒出芽时从宿主细胞膜上获得的结构[12]。在病毒外包膜上能检测到pEP402R(CD2v)蛋白存在。病毒外包膜的存在一方面能保护病毒粒子,另一方面使病毒入侵细胞和出芽过程复杂化,但外包膜的存在并不影响病毒的感染性[10]。
病毒衣壳的功能主要是保护病毒核酸并参与病毒感染过程。ASFV 衣壳最大直径为250 nm,呈一个六边形的对称立体结构[13]。P72 是主要的病毒衣壳蛋白。有研究[10]表明,病毒二十面体衣壳主要由8 280 拷贝的p72 蛋白和其余少量衣壳蛋白(如pE120R 和pB438L 蛋白)组成,其中p72 约占病毒颗粒总重量的32%。
内膜是由内质网分离出来的厚约70Å 的脂质双分子层结构[14]。在ASFV 内膜上存在p17、pE183L、p12、pE248R、pH108R 和pE199L 蛋白,其中p17 和pE183L 主要作用是帮助组装病毒衣壳,p12、pE248R 和pE199L 则参与病毒入侵宿主细胞[4]。
核壳是病毒的一个独立结构域,直径为180 nm,主要由多聚蛋白前体pp220 和pp62组成[15]。pp220和pp62均含有多个蛋白酶水解位点,其中,Gly-Gly-Xaa 位点可以被特异性的SUMO-1半胱氨酸蛋白酶pS273R 识别并切割,pp220 被水解成p5、p14、p34、p37 和p150,pp62 则被水解成p8、p15 和p35,水解后的蛋白为成熟蛋白,这一过程对于ASFV 粒子成熟和感染性具有重要意义[10]。pp220 与pp62 之间存在密切的交互应答作用,pp62 的加工依赖于pp220 的表达,而pp62 对pp220 的亚细胞定位有重要影响,pp62 和pp220缺失会影响病毒粒子类核发育状态,产生空核粒子[15-16]。
ASFV 粒子核心部位是核壳包裹着的类核,其包含ASFV 基因组,编码病毒结构蛋白和与病毒复制、转录和免疫抑制相关的非结构蛋白。pA104R是存在于类核处的一种重要DNA 结合蛋白,对ASFV 基因的包装和复制至关重要。pA104R 以14~16 nt/bp 的速率与单链DNA(ssDNA)和双链DNA(dsDNA)结合,与ASFV 拓扑异构酶II pP1192R 共存时显现出DNA 超卷曲活性。由于pA104R 在ASFV 复制中发挥关键作用,因此它是目前对抗ASFV 感染的一个重要靶点[10]。
2 ASFV 的生命周期
2.1 吸附和入侵
内吞作用是许多病毒用来通过细胞膜物理屏障进入细胞的一种方式。病毒内吞进入细胞依赖于特定的细胞信号通路激活,并由病毒和细胞相互作用所驱动[17]。早期研究表明,ASFV 入侵宿主细胞依赖于低pH、温度、能量和胆固醇[18],并且由于ASFV 对细胞具有趋向性,因此认为巨噬细胞的某些受体(CD163、CD45、MHC-II 等)可能介导了病毒入侵这一过程。然而,基因敲除猪的感染试验结果[19]表明,CD163 对于ASFV 感染并非是必需的,还有其他未明确的特异性受体蛋白参与这一过程。
近些年的研究[20]主要认为,ASFV 通过网格蛋白介导的内吞作用(clathrin-dependent endocytosis,CME)和巨胞饮作用进行内化。在CME 过程中,病毒颗粒首先与细胞膜表面的特定受体结合,受体接受信号后,膜上相应部位会形成网格蛋白包被小窝,随后形成直径85~120 nm 的包被囊泡,而病毒粒子就被包裹在包被囊泡中[17]。囊泡形成后,会在GTPase 动力驱动下从细胞膜脱落并进入细胞质,此时囊泡失去网格蛋白,形成早期内体并进一步成熟[21]。而在巨胞饮过程中,无需受体介导,主要由多种激酶,如RTK、PI3K1、PAK1 等和Rho GTPases 激活而在质膜上产生褶皱或泡状结构来完成。通过巨胞饮作用入侵细胞的病毒粒子被包裹在0.5~10 mm 的无包被的囊泡中,形成巨胞饮体[22]。在形成早期内体或巨胞饮体后,ASFV 粒子会被进一步运输到晚期核内体,进行病毒脱壳。随后ASFV 内膜与晚期内体膜之间发生融合,将病毒类核释放到细胞质中,并在细胞质中形成病毒工厂(virus factory,VF)进行复制[2]。
2.2 基因表达和复制
ASFV 基因表达有严格时间调控,可以分为早期、中期和晚期3 个阶段,其中早期基因表达先于ASFV DNA 的复制[23]。早期基因表达发生在感染后4~6 h,ASFV 的RNA 聚合酶启动早期基因表达,产生病毒复制所需的蛋白质。ASFV 基因组的复制也分为两个阶段,最初发生在细胞核中,随后转移到细胞质VF 中进行。感染后6~8 h,ASFV 通过G1211R基因编码的自身DNA 聚合酶启动复制。在感染8~16 h 时,开启中晚期基因表达,产生病毒结构蛋白,随后被组装成病毒粒子[2,4]。
2.3 组装和出芽
VF 定位于靠近细胞核的细胞质中,它既是ASFV 基因组复制的主要区域,也是子代病毒组装的场所[4]。ASFV 内膜前体的形成启动了子代病毒组装过程,跨膜结构蛋白p54 在内质网上负责膜的募集和转化。内膜前体形成后,沉积在膜组装中间体上的衣壳蛋白(p72 和其伴侣蛋白pB602L等)会逐个拼装在一起,使膜前体逐渐形成二十面体结构[24]。伴随着衣壳形成,核壳在病毒内膜下进行组装,多蛋白加工酶pS273R 在这一过程中发挥重要作用,pB602L 和p17 也通过影响pp220 和pp62 的水解来影响核壳到内膜的组装[25]。最后进行基因组和相关类核蛋白包装,主要由位于核壳的pA104R 发挥作用[26]。病毒组装完成后,在微管和驱动蛋白的共同作用下从被感染细胞中出芽,病毒衣壳蛋白pE120R 促进成熟病毒颗粒从组装位点运输到质膜,在质膜中病毒获得外包膜。总体来说,一个完整的ASFV 感染周期,从附着细胞到出芽在24 h 内完成[2]。
3 ASFV 对先天性免疫反应的逃逸
病毒入侵机体后,先天性免疫反应是抵御病毒的第一道防线,由上皮细胞或巨噬细胞产生干扰素(interferon,IFN)及其他细胞因子,进而诱导自然杀伤细胞、粒细胞和树突状细胞激发出显著的主要的先天性免疫反应,并刺激启动获得性免疫反应,包括B 细胞发挥的特异性体液免疫反应和T细胞发挥的适应性细胞免疫应答,进而对病毒进行彻底清除并建立免疫记忆[4,27]。
在先天性免疫反应中,IFN 是重要组成部分,其可诱导细胞内抗病毒蛋白的表达从而干扰病毒生命周期,并抑制病毒在细胞间扩散[28]。当病毒感染宿主细胞时,各种模式识别受体(pattern recognition receptors,PRRs),识别病原体相关分子模式从而激活先天性免疫信号通路,产生炎症细胞因子和IFN-Ⅰ[29]。为保证自身的生存和复制,ASFV 进化出多种对抗宿主细胞免疫反应的策略,其中包括抑制感染细胞中IFN 和IFN 刺激基因(IFN-stimulated genes,ISG)表达,以及抑制肿瘤坏死因子α(tumor necrosis factor,TNF-α)、白细胞介素(interleukin 包括IL1-β、IL-6、IL-8)等炎症因子释放[9]。本文将从IFN 反应和炎症反应两个方面来阐述ASFV 对先天性免疫反应信号通路的调控,其分子作用网络如图1 所示。
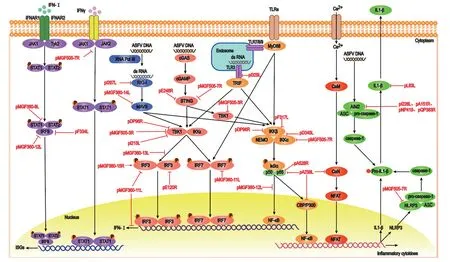
图1 ASFV 拮抗宿主IFN 反应及炎症反应的作用网络
3.1 ASFV 对IFN 反应的调节
ASFV 基因组为DNA,因此细胞质DNA 传感器在病毒识别和IFN 激活中发挥重要作用。具体而言,环二核苷酸合成酶(cyclic GMP-AMP synthase,cGAS)可识别病毒DNA,发生去谷氨酰化和类泛素蛋白修饰进而活化,活化的cGAS会催化第二信使分子环二核苷酸(cyclic GMPAMP,cGAMP)合成,进而cGAMP与干扰素刺激因子编码蛋白(stimulator of interferon,STING)结合。cGAMP 与STING 的相互作用诱导STING 形成二聚体,进而与iRhom2 蛋白结合并易位至ER-golgi 中间区室(ERGIC),与TANK 结合激酶1(TANK binding kinase 1,TBK1)结合,TBK1 对干扰素调节因子3(interferon regulatory factors 3,IRF3)进行磷酸化,磷酸化的IRF3 入核并介导IFN-Ⅰ表达[8,30],此外,STING 与TBK1的激活还会激活IKK 复合体(inhibitor of NF-κB,IκB,包含IKKβ、IKKα 和NEMO)从而将NF-κB的抑制因子(inhibitor of NF-κB,IκB)磷酸化以及蛋白泛素化和降解,释放转录因子NF-κB 入核并介导IFN-Ⅰ和前炎性因子表达[31-33]。除了cGASSTING 信号轴以外,在III 型RNA 聚合酶(RNA polymerase III,RNA PolyIII)作用下,两种PRRs包 括Toll 样受体(Toll-like receptors,TLRs)和RIG-I 样受体(RIG-I-like receptors,RLRs)也发挥了病原体识别和IFN 激活功能[34]。细胞分泌的IFN 可被IFN 受体复合物IFNAR1/ IFNAR2 二聚体识别,并招募胞内的酪氨酸激酶TYK2 和JAK1 到细胞膜并进行相互磷酸化,进而STAT1 和STAT2被分别招募和磷酸化,并与IRF9 形成三元复合物IFN 刺激基因因子3(ISGF3),ISGF3 入核与IFN 刺激反应元件(ISREs)结合,启动ISGs 转录,产生的ISGs 可直接阻遏病毒复制和扩散,清除病毒[9,35]。
ASFV 编码多种蛋白质来抑制IFN-Ⅰ的生成,主要分为两大类:一类是多基因家族蛋白(MGF360、MGF505),另一类是其他蛋白(pI329L、pDP96R、pE120R、pI215L、pF334L、pI267L)。ASFV 编码蛋白对IFN 产生的分子调控机制研究总结如表1 所示。

表1 ASFV 编码的干扰素抑制相关蛋白及具体作用机制
在ASFV 的多基因家族中,含有11~15 个成员的MGF360 和含有9~10 个成员的MGF505 被证明可以抑制IFN-I 反应[36-37]。研究表明,在MGF360家族中,pMGF360-12L 以核定位信号依赖的方式与KPNA2、KPNA3 和KPNA4 竞争结合NF-κB,从而阻断NF-κB 的核易位,抑制IFN-β 表达[38],还能通过降低IRF9 总蛋白水平抑制IFN-β 介导的免疫效应[39];pMGF360-15R 能够以NF-κB 非依赖性的方式靶向IRF3,调节IFN-β 表达,但不影响I 型或II 型IFN 对Janus 激酶信号传导及转录激活因 子(Janus-activated kinase Singal transducers and activators of transcriprion,JAK-STAT)通路的激活[40];pMGF360-13L 能抑制cGAS-STING 通路介导的IFN-β 活化,拮抗TBK1 和IRF3 向STING 募集,还能抑制TBK1 和IRF3 磷酸化,以及IRF3二聚化和入核,从而调控宿主的IFN 反应[41];pMGF360-9L 与STAT1 和STAT2 存在相互作用,并通过泛素-蛋白酶体对二者进行降解,抑制IFN抗病毒效应通路激活[42]。pMGF360-11L 能抑制cGAS、STING、TBK1、IKKε、IRF7 和IRF3-5D介导的IFN-β 和ISRE 启动子的激活,与TBK1 和IRF7 相互作用,通过半胱氨酸、泛素-蛋白酶体和自噬途径降解TBK1 和IRF7,还能抑制TBK1和IRF3 磷酸化,通过负向调控cGAS 信号通路参与IFN-I 表达[43];pMGF360-14L,可能通过竞争结合MAVS,抑制TRIM21 对MAVS 的泛素化,抑制IFN-I 产生[44]。而在MGF505 家族中,有研究表明MGF505-3R 与cGAS/TBK1/IRF3 相互作用,靶向TBK1 从而破坏了cGAS-STING 介导的IFN-β产生信号通路[45];pMGF505-7R 通过与STING 和自噬启动因子丝氨酸苏氨酸激酶Unc-51 样自噬激活激酶1(ULK1)相互作用,导致STING S366 被ULK1 磷酸化,并通过自噬途径降解STING,从而抑制cGAS-STING 途径介导的IFN-I 表达[8];除了抑制IFN-I 之外,pMGF505-7R 还能抑制IFN-γ 介导的下游基因转录,并与JAK1 和JAK2 相互作用,破坏IFN-γ 信号通路[8,46]。
除了多基因家族以外,ASFV 还编码一些其他蛋白质来抑制IFN 反应。pI329L 是TLR3 的同源物,通过靶向TLR3 通路中的TRIF,竞争性地结合到TLRs 的下游信号分子上,从而影响IFN 产生[47-48];pDP96R 是ASFV 的一个毒力因子,通过cGAS-STING 信号通路负调控IFN-I 产生,并通过阻断TBK1 和IKK-β 的激活影响NF-κB 信号传导[49];pD345L 对于cGAS-STING 诱导的IFN-β 和NF-κB 激活有抑制作用,与IKKα 以及IKKβ 互作,破坏激酶活性,抑制IκBα 磷酸化[50]。pE120R 是一种与病毒转运相关的蛋白,它与IRF3 羧基末端结构域相互作用,干扰IRF3 向TBK1 募集,进而抑制IRF3 的磷酸化,通过cGAS-STING 通路抑制IFN-β 生成[51];pI215L 是病毒的一种E2 泛素结合酶,能抑制TBK1 K63 泛素化,从而抑制IFN-β 产生[52]。pF334L 通过与IRF9 相互作用,诱导IRF9通过泛素-蛋白酶体途径发生降解,阻碍ISGF3的部分入核,抑制ISGs 分子表达[53]。pI267L 通过与E3 泛素连接酶RIPLET 相互作用中断其与RIG-I 的联系,从而损害RIPLET 介导的泛素化和RIG-I 的激活,抑制IFN-β 生成[35];pA528R 能与p65 蛋白相互作用,抑制TLR8-NF-κB 通路及其介导的抗病毒反应[54];pE248R 能招募ATG101 降解STING,通过负调控cGAS-STING 信号通路抑制IFN-I 产生[55]。总之,为逃避宿主细胞的免疫反应,ASFV 编码多种蛋白质以不同策略抑制IFN 产生,从而保证自身存活和复制,提高感染效率。
3.2 ASFV 对炎症反应的调节
炎症反应在ASFV 的发病机制中也发挥着重要作用。ASFV 感染猪后,会诱导巨噬细胞分泌多种炎性细胞因子,如TNF-α、白细胞介素(IL-1β、IL-6、IL-8)、CCL4、CXCL10 等,引起强烈炎症反应,导致受感染猪发生严重的炎症病变甚至死亡[9]。有研究[5]发现,与高毒力ASFV 毒株相比,低毒力毒株能诱导巨噬细胞产生更高水平的炎性细胞因子,说明炎症反应与ASFV 感染致病力密切相关。ASFV 编码蛋白对炎性因子产生的分子调控机制研究总结如表2 所示。

表2 ASFV 编码的抑制炎症反应相关蛋白及功能
NF-κB 信号通路是机体通过炎症反应进行抗病毒的重要途径。细胞中的PRR 可识别病毒DNA或dsRNA 激活NF-κB通路,使NF-κB二聚体入核,在细胞核中积累并调控基因转录[56]。炎症反应的发生,除了通过对NF-κB 通路的激活,还能通过激活NLRP3 炎症小体途径,促进炎症因子分泌[57]。
ASFV pA238L、pF317L、pL83L 和pMGF505-7R 蛋白参与细胞炎症反应调控。pA238L 蛋白对炎症因子转录活动的调控有多种途径:首先它是IκB的同源物,可以与NF-κB 结合抑制NF-κB 激活,从而抑制细胞的炎症反应;其次它能与钙调神经磷酸酶(calcium/calmodulin-regulated phosphatase calcinerurin,CaN)相互作用,影响其磷酸酶活性,抑制活化T 细胞核内因子(nuclear factor of activated T cell cytoplasmic,NFAT)激 活,抑制IL-2、IL-4 等细胞因子产生;最后pA238L 能与细胞核中CBP/p300 相互作用,破坏p300 向转录复合体的募集,阻止p300 磷酸化,进而抑制TNF-α产生[9,58]。研究[59]发现,在ASFV 感染的细胞中,pF317L 与IKKβ 相互作用,抑制IKKβ 磷酸化,导致IκBα 磷酸化和泛素化降低,IκBα 表达上调,从而抑制NF-κB 激活,抑制细胞炎症反应。
作为一种重要的促炎细胞因子,IL-1β 由单核-巨噬细胞分泌后可引起炎症反应并促进B 细胞增殖分化[4]。然而研究[60]表明,ASFV pL83L 蛋白可特异性结合IL-1β,从而阻断IL-1β 与受体结合,抑制IL-1β 引起的炎症反应。pMGF505-7R 是一个多功能蛋白质,与IKKα 相互作用,抑制IκBα磷酸化和NF-κB 核易位,从而抑制IL-1β 前体转录。另外,pMGF505-7R 还与NLRP3 相互作用,阻断NLRP3 炎症小体组装,抑制成熟IL-1β 分泌[57,61]。除此之外,还有研究[62]发现pI226L、pA151R、pNP419、pQP383R 蛋白能抑制AIM2 炎症小体组装,从而抑制IL1-β 成熟和分泌。
4 ASFV 对适应性免疫反应的逃逸
近些年来,研究[9]表明,用ASFV 弱毒株感染猪可以产生保护性抗体,且弱毒株免疫血清能够提高健康猪对同源毒株的抵抗力,说明宿主细胞的适应性免疫在抵抗ASFV 感染中也发挥着重要作用。在抗原递呈和T 细胞免疫反应中,巨噬细胞的MHC-I 类和MHC-II 类分子发挥关键作用。在ASFV 感染过程中,其编码的pEP402R、pEP153R蛋白能通过不同的分子机制来逃避适应性免疫反应。
pEP402R 蛋白也称为CD2v 蛋白,是ASFV感染晚期编码的一种蛋白质,存在于ASFV 粒子外包膜上,其结构和功能与T 淋巴细胞表面抗原CD2 相似,它能介导病毒与红细胞粘附,促进病毒在宿主体内传播[63-64]。除此之外,CD2v 可以通过与淋巴细胞表面受体结合,或者通过与细胞质蛋白之间相互作用抑制淋巴细胞增殖,从而抑制适应性免疫[48]。研究[63]表明,CD2v 缺失的BA71 减毒活疫苗对不同基因型毒株可产生一定保护效果。
pEP153R 蛋白则与一些细胞蛋白同源,如CD94、CD69、Ly94A 和CD44 等,因此pEP153R能与这些细胞蛋白竞争结合MHC 类分子,从而抑制T 细胞激活[65];pEP153R 与MHC 类分子的结合也会抑制MHC-I 从内质网到细胞膜的运输,抑制NK 细胞激活,由此可见pEP153R 蛋白也在抑制适应性免疫方面发挥关键作用。
5 ASFV 对程序性细胞死亡进行调节
病毒入侵时,机体会启动应激性细胞行为以控制病毒感染,例如细胞程序性死亡(细胞凋亡和细胞自噬等),这是宿主抗病毒免疫应答的重要部分。然而,ASFV 在感染过程中会通过病毒蛋白来调控细胞程序性死亡的发生,达到逃避宿主免疫反应并维持自身复制的目的。
细胞凋亡主要有3 种途径,分别为外源性凋亡途径(extrinsic apoptotic pathway,EAP)、内源性凋亡途径(intrinsic apoptotic pathway,IAP)和内质网应激诱导途径。EAP 由胞外的凋亡受体介导与相关死亡结构域以及procaspase-8 形成诱导死亡信号复合体,从而依次激活caspase 8 和caspase 3 进行细胞凋亡。IAP又称为线粒体/细胞色素C(cytochrome C,Cyt-c)介导的通路,在其介导的细胞凋亡中,细胞内刺激因子与线粒体上的Bcl-2 家族成员相互作用,将Cyt-c 从线粒体释放到细胞质中,Cyt-c、APAF-1 和caspase 9 形成凋亡小体,激活caspases9/3/6/7,从而引起凋亡[4]。内质网应激诱导途径则能通过持续内质网应激启动未折叠蛋白反应,导致caspase-12 活化,从而导致凋亡[66]。此外,细胞自噬也是细胞的一种防御保护措施[67],病毒入侵宿主细胞后,会通过病毒蛋白诱导、病毒配体与受体互作、内质网应激3 种机制来诱导宿主细胞发生自噬,将病毒颗粒运送到溶酶体中进行降解,并进一步通过与PRRs 结合,诱导IFN 反应[68-69]。宿主细胞通过凋亡或自噬,能够使病毒暴露在免疫系统中,从而抑制病毒复制进而将病毒清除。因此为确保感染早期细胞的存活状态,产生足够的子代病毒,病毒进化出了延迟或者抑制细胞程序性死亡的策略,进一步为确保感染晚期子代病毒的成功出芽,病毒还进化出了促进细胞程序性死亡的策略[9]。
在ASFV 编码蛋白中,pA224L、pA179L、EP153R 和pDP71L 发挥抑制凋亡功能,P54/pE199L 发挥诱导细胞凋亡功能。其中,pA179L 还参与细胞自噬调控。
pA224L 是在ASFV 感染晚期表达的一种保守蛋白质,它是一种凋亡抑制因子(inhibitor of apoptosis proteins,IAPs)同源蛋白,能够与激活的caspase-3 相互作用并抑制其蛋白酶功能,从而抑制多种凋亡诱发因子诱导的细胞凋亡[9,70]。另有研究表明,pA224L 可通过IKK 激活转录因子NF-κB 从而抑制细胞凋亡[71],然而,A224L 的缺失对ASFV 毒力以及感染细胞的细胞凋亡程度并无显著影响[72]。
pA179L 定位于线粒体或内质网,是Bcl-2 家族的同源物,它含有BH1、BH2、BH3 和BH4 的保守结构域,但缺乏相应跨膜结构域。结构生物学结果[9,73]显示,pA179L 既能结合促凋亡蛋白Bid的活性截断体p13 和p15 从而抑制其活性,也能结合其他促凋亡蛋白Bak 和Bax 实现细胞凋亡的抑制。最近研究[74]发现,pA179L 能够发生异戊二烯化修饰,异戊二烯化通过指导pAl79L 蛋白的线粒体定位调控其抑制细胞凋亡活性。进一步研究[75]表明,pAl79L 蛋白通过其BH3 同源结构域与Beclin-1 在线粒体和内质网相互作用,在饥饿条件下抑制自噬体形成,这提示pAl79L 蛋白具有对程序性细胞死亡的多重调控功能,以保障病毒复制和子代病毒组装。
EP153R 是C 型凝集素类似蛋白,参与病毒感染后的血细胞吸附过程,还能抑制caspase-3,除此之外它还能降低细胞蛋白p53 的反式激活活性,从而某些凋亡抑制分子的转录活动受到影响,抑制细胞凋亡[76]。
内质网应激也是细胞凋亡发生的途径之一,而在ASFV 复制过程中,内质网中积累过量的未折叠蛋白或错误折叠蛋白,则会诱导未折叠蛋白反应(unfolded protein response,UPR)。而ASFV可选择性活化ATF6 信号通路,激活caspase-12,诱导分子伴侣钙联蛋白和钙网状蛋白表达,改善内质网应激状态,抑制感染早期细胞凋亡活动[77]。pDP71L 是ASFV 中高度保守的晚期蛋白,它是单纯疱疹病毒ICP34.5 以及细胞蛋白GADD34 的类似物,均可与蛋白磷酸酶PP1 结合,并招募真核翻译起始因子eIF2α 去磷酸化,阻断ATF4 和下游促凋亡因子CHOP 生成,从而阻断内质网应激诱发细胞凋亡的信号轴eIF2α-ATF4-CHOP[78],保障细胞的蛋白质翻译功能以及病毒增殖。然而pDP71L缺失体病毒研究结果[79]显示,该蛋白并不是调控eIF2α 的唯一因素。
除抑制细胞凋亡以外,ASFV 还可以通过相应策略促进细胞凋亡,有利于病毒传播,从而增加病毒从细胞中释放的量,避免炎症信号的诱导[9]。p54(E183L)位于成熟病毒粒子内膜,是一种结构蛋白,在感染早期参与吸附宿主细胞,并通过自身的SQT 基序(与BCL2 家族的促凋亡蛋白BH3-only BIM 同源)与微管动力蛋白直接结合,实现病毒内化后向病毒复制工厂的转运。在病毒感染后期p54 高量表达,激活caspase-3 并诱导细胞凋亡,且将内质网膜转化为子代病毒内囊膜。研究[80]表明,SQT 基序内结构域的缺失可使P54 蛋白丧失与微管动力蛋白结合的能力以及诱导细胞凋亡的能力。pE199L 是在感染晚期表达的一种内膜蛋白,能够激活促凋亡因子Bak 从而诱导细胞凋亡[81]。因此为提高感染率,ASFV 通过不同策略对宿主细胞的凋亡程序进行调控。
6 总结与展望
自2018 年我国首次报告ASF 病例以来,ASF给我国乃至全世界都带来了巨大经济损失。ASFV在感染宿主细胞过程中,细胞会识别病毒刺激,引起IFN、炎症、程序性细胞死亡和机体适应性免疫反应,以形成系统的抗病毒反应。同时,ASFV 进化出一系列逃逸宿主细胞抗病毒反应的策略,包括调节先天性免疫信号通路、抑制抗原呈递、调控程序性细胞死亡等,从而躲避细胞的免疫攻击,提高感染效率。以上复杂的免疫逃逸策略加大了有效疫苗设计的难度。
基于对ASFV 编码蛋白的鉴定和功能研究,已证实p72、p30、p54 和CD2v 等蛋白是ASFV 的重要免疫原,可诱导猪体产生中和抗体,然而免疫保护效果一般[82]。在缺失体病毒构建工作中发现,DP71L、DP96R、B119L 或DP148R 的缺失可减弱病毒毒力,有一定保护性效果[83]。目前,国内外对ASFV 疫苗的设计开发以重组亚单位疫苗和减毒活疫苗为主,以上免疫原和减毒策略的尝试并未达到理想的免疫保护效果。有研究[84-85]表明,7 基因缺失的减毒活疫苗HLJ/1-7GD(MGF505-1R、MGF360-12L、MGF360-13L、MGF360-14L、MGF505-2R、MGF505-3R和CD2v缺失)有较好的临床免疫保护效果,但该疫苗株的生物安全性还有待于进行全面系统评估,因此进一步对ASFV 编码产物的鉴定及其功能进行充分研究,可为重组减毒活疫苗的基因缺失策略提供更多缺失基因组合,有助于构建具有良好免疫原性和强保护效果的疫苗株[86]。此外,通过分子流行病学对ASFV 基因组进化的监测发现,有2 株猪场分离毒株发生了缺失并导致病毒毒力减弱,可能导致猪群的慢性或无症状感染,鉴定结果表明缺失基因为pEP402R和pEP153R[87],回顾二者编码蛋白的功能研究,可发现它们参与宿主适应性免疫反应的逃避或调控细胞程序性死亡。因此,在未来科学防治和新疫苗开发中可考虑当前ASFV 的适应和进化。
已有研究[88-89]借助于生物信息学研究手段挖掘ASFV 免疫逃逸相关蛋白或基于T 细胞表位全景扫描技术开发ASFV 细胞免疫疫苗,这些工作为ASFV 疫苗研发工作带来了新突破口。未来,除了对IFN 生成和炎症反应调控的分子机制鉴定外,还应结合适应性免疫中免疫细胞亚群偏移和病理性炎性反应发生的机理研究,为新型疫苗设计提供理论依据,加强免疫保护效果。
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09 06:12:12
科学(2020年3期)2020-11-26 08:18:22
汉字汉语研究(2020年2期)2020-08-13 07:52:48
当代水产(2020年3期)2020-06-15 12:03:02
电子制作(2019年22期)2020-01-14 03:16:24
疯狂英语·新读写(2018年3期)2018-11-29 22:37:11
实用皮肤病学杂志(2015年4期)2015-12-22 11:21:42
中国病理生理杂志(2015年8期)2015-12-21 12:38:06
医学研究杂志(2015年12期)2015-06-10 06:57:46
医学研究杂志(2015年3期)2015-06-10 06:41:52
