
构筑三维有序介孔少层MoS2/C复合材料及其电化学析氢性能
2022-12-07 12:29李博文余宗宝杨占旭
燃料化学学报 2022年10期
李博文,韩 乔,余宗宝,杨占旭
(辽宁石油化工大学 石油化工学院,辽宁 抚顺 113001)
随着人类生活水平的提高,对能源的需求也日益增加。由于燃料化石能源的不可再生性和对环境的污染,开发清洁可持续的能源已经刻不容缓。H2作为一种高效、可持续的清洁能源具有良好的开发前景和商业价值[1]。电解水制氢实现了原料和产物的循环利用,全过程无污染。铂族贵金属催化剂是目前公认最佳的电化学析氢催化剂,但由于储量少且成本高,限制了其大规模的应用[2,3]。过渡金属硫化物凭借其独特的结构和电子特性成为广受关注的非贵金属催化剂[4,5]。
MoS2作为过渡金属硫化物因其边缘对电化学析氢反应(HER)具有高效的催化活性,所以被广泛研究[6]。目前,主要通过制造缺陷[7-9]、杂原子掺杂[10-12]、或构建异质结构[13-15]等方法调控MoS2的电子排布或暴露更多的MoS2边缘活性位点,从而提高MoS2的催化活性。Wang等[16]采用H2O2化学刻蚀法,在MoS2纳米片表面均匀引入单个S原子空位,通过控制刻蚀条件,确定单一S原子空位的密度和分布,从而得到最佳析氢活性,电流密度为10 mA/cm2时,过电位为131 mV,塔菲尔斜率为48 mV/dec。Ge等[17]通过在MoS2上引入的电负性更强的O杂原子,激活了MoS2表面的惰性基面,为HER提供了大量的活性位点,提高了电传输效率,并且诱导MoS2从2H相转变为导电性更好的1T相。在碱性电解液中,制备的O-MoS2在10 mA/cm2时过电位仅为120 mV,Tafel斜率为84 mV/dec。密度泛函理论计算表明,在MoS2中引入O杂原子优化了对H*的吸附,降低了水解离的吉布斯自由能。Zhao等[18]通过水热法构建了花状的MoS2/NiS异质结构HER催化剂,实现了电子的快速转移并暴露了更多的活性位点,在碱性条件下过电位为158 mV且具有良好的稳定性。以上方法虽然有效地提高了MoS2的催化活性,但是一维或二维结构的催化剂存在一定的堆叠、团聚的情况,降低了催化剂的比表面积,活性位点没有充分的暴露[19]。
而构建三维结构的MoS2电化学析氢催化剂可以有效的避免以上问题。三维结构中的孔道可以为反应提供更大的比表面积,使活性位点充分暴露在电解液中,并为物质传输和电子转移提供了通道[20]。还可以将活性组分直接构建在3D的导电基底上(如碳基底[21]),这可以显著提高电子转移速率。凭借以上优势构建三维结构MoS2电化学析氢催化剂受到人们的广泛研究。Meng等[22]通过牺牲SiO2小球合成了具有蜂窝状三维介孔MoS2和石墨烯杂化结构HER催化剂,暴露了MoS2丰富的边缘位点并提升了导电性,进一步掺杂Co原子后,在0.5 mol/L H2SO4溶液中,电流密度为10 mA/cm2时,过电位为143 mV。Zhai等[23]以NiMoS结构为前驱体,通过氧等离子体氧化处理和随后的氢化调节,将二维(2D) MoOx/MoS2纳米片与一维(1D) NiOx/Ni3S2纳米棒相结合,制备了三维(3D) NiMoOx/NiMoS异质结构催化剂,在电流密度为10 mA/cm2时,HER的过电位为38 mV,OER的过电位为186 mV。Shi等[24]以介孔硅材料为模板采用纳米铸造的和高温H2S还原硫化的方法,制备了一系列具有特定结构的过渡金属硫化物。Lu等[25]以SBA-15为硬模板制备了N掺杂CMK-3,通过浸渍和热还原的方法将非晶MoS2分散在N-CMK-3表面,以N-CMK-3的介孔结构限制了MoS2的团聚,在电流密度为10 mA/cm2时,HER的过电位为184 mV。Zhu等[26]以SBA-15为模板,甲硫氨酸与金属氯化物为前驱体,通过无溶剂纳米铸造的方法合成了一系列具有不同结构的过渡金属硫化物与氮掺杂碳复合介孔材料,其中,OM-MoS2@NC-900-5在电流密度为10 mA/cm2时,HER的过电位为218 mV。
尽管已经有很多关于纳米铸造法制备三维结构催化剂的研究,但前驱体的选择对制备催化剂的性能和安全环保也有着巨大的影响,所以寻找更廉价、环保的前驱体制备高比表面积以及MoS2活性位点高度分散的HER催化剂始终存在挑战。本实验通过设计三维结构催化剂,以SBA-15为硬模板,蔗糖为碳源,四硫代钼酸铵(ATTM)为MoS2前驱体,采用纳米铸造法获得高度有序介孔三维结构,以实现材料的高比表面积,通过简单的煅烧使ATTM原位分解生成MoS2,利用碳限制MoS2的团聚以生成少层MoS2薄片,通过调节MoS2与碳的比例,实现对MoS2分散和边缘暴露程度的控制,从而制备出大量暴露MoS2薄片边缘的三维MoS2/C-SBA-15复合材料电化学析氢催化剂。
1 实验部分
1.1 试剂与仪器
聚环氧乙烷-聚环氧丙烷-聚环氧乙烷三嵌段共聚物(P123,MW = 5800,EO20PO70EO20)购自Sigma-Aldrich;正硅酸乙酯99% (GC)、钼酸铵((NH4)6Mo7O24·4H2O,AR)、硫化铵溶液((NH4)2S,20%in H2O,AR)和氢氟酸(HF,40% in H2O,AR)购自阿拉丁化学试剂有限公司;蔗糖(C6H22O11,AR)购自国药集团化学试剂有限公司;无水乙醇(C2H5OH,AR)、氨水(H5NO)购自天津市大茂化学试剂厂;碳纤维纸(CFP,TGP-H-060) 购买自日本东丽公司。硫酸(0.5 mol/L)、盐酸(2 mol/L)自制。所有实验均使用去离子水。
电子分析天平(AUW120D),日本岛津公司;真空管式加热炉(HTL1100-60),上海皓越仪器设备有限公司;电热恒温水浴锅(XMTD-400),北京市永光明医疗仪器;电热鼔风干燥箱(GZX-9070MBE),上海博讯实业有限公司医疗设备厂;数显电动搅拌器(B11-2),上海司乐仪器有限公司;电化学工作站(VSP-300),Biologic。
1.2 材料的制备
高温水热条件下合成了有序介孔二氧化硅硬模板SBA-15,高温水热条件增加了SBA-15壁的中孔道数量,有利于后续制备复合材料[27]。通过钼酸铵与硫化铵制备四硫代钼酸铵(ATTM)[28]。
MoS2/C-SBA-15复合材料的制备:取0.736 g蔗糖和0.492 g ATTM完全溶解于5 mL去离子水中,随后加入1 g硬模板SBA-15,充分搅拌,室温浸渍24 h。将混合物转移至电热鼔风干燥箱中100 ℃干燥6 h,再加热至160 ℃保温6 h。将固体研磨得到黑色粉末。再取0.492 g蔗糖和0.328 g ATTM溶于5 mL去离子水中,与黑色粉末充分混合,重复上述步骤,得到黑色固体转移至管式炉中,在H2含量5%的H2/N2混合气气氛下,以 2 ℃/min的升温速率从室温加热至850 ℃并保温4 h。随后样品经自然降温、研磨。将得到的黑色粉末加入到HF(10%,50 mL)溶液中搅拌12 h,以除去硬模板SBA-15。通过离心分离、水洗、乙醇洗并60 ℃真空干燥6 h,得到黑色样品。通过改变蔗糖与ATTM的投料质量比,制备不同MoS2含量的样品,命名为MoS2/CX-SBA-15,多个样品表示为MoS2/C- (X-Y) -SBA-15。其中,X、Y为投入ATTM的质量分数(%)。具体制备过程如图1所示。
1.3 表征
采用X射线衍射仪(德国 Bruker D 8 Advance)测试样品材料的物相组成及有序介孔结构。测试条件为:CuKα 衍射源,所需管电压和管电流分别40 kV和20 mA。采用扫描电子显微镜(日本日立Hitachi SU8010)对材料的表面形貌进行观察表征,测试工作电压为 15 kV。采用透射电子显微镜(日本电子 JEM-2100)进行样品微观形貌观察,同时用高分辨透射电镜照片(HRTEM) 和选区电子衍射谱(SAED) 分析其微观结构与形态。采用X 射线光电子能谱仪(ESCALAB 250)表征分析元素的键合结构和电子价态。测试条件为:激发源为 AlKα X 射线源 (1482.6 eV),工作电压为 12 kV,功率为120 MW,粒度为 380 目,切割面积为 6 mm × 8 mm,负载量为 100 mg。拉曼光谱使用美国赛默飞DXR拉曼显微镜测试获得。工作具体条件为:波长为532 nm的高亮度激光源激发,采集曝光时间 1.0 s,预览采集时间 0.5 s,样品与背景曝光均为 100 s。氮吸附-脱附表征测试使用的是 Autosorb-IQ2-MP自动比表面积及孔径分布实验设备,并以Brunauer-Emmett-Teller(BET) 为计算的理论依据,计算得到所合成材料的比表面积与材料各类孔径的分布情况。
1.4 电化学性能测试
将 0.01 g 样品分散到 900 μL 无水乙醇与 100 μL Nafion 溶液 (质量分数5%) 的混合溶液中,超声处理 20 min,将300 μL的混合溶液分六次均匀滴在碳纸两面,干燥凝固后得到测试电极。所有电化学性能测试均使用 VSP-300电化学工作站在标准三电极体系下进行,以0.5 mol/L H2SO4溶液为电解液,选用石墨电极为对电极,饱和甘汞电极(SCE)为参比电极。线性扫描伏安法 (LSV) 扫描速率为2 mV/s,最小电压为 0 V,最大电压为-1.0 V;电化学阻抗谱 (EIS)测试频率为 0.1-100 MHz。采用计时电位法在电流密度为 10 mA/cm2的条件下对材料进行稳定性测试。测量前,电极均以 100 mV/s 的扫描速率进行循环伏安扫描20 圈。电位用以下等式校准到可逆氢电位 (RHE):
2 结果与讨论
2.1 MoS2/C-X-SBA-15复合材料的组成结构和形貌分析
通过扫描电镜(SEM)对合成的样品进行表征,如图2所示,SBA-15呈短六角形柱状,短柱两端呈阶梯状,粒径约为1 μm,MoS2/C-(1-40)-SBA-15完美的复制了SBA-15的形貌,且可以清楚观察到表面的孔道结构。ATTM投料比较低时,MoS2/CX-SBA-15整体结构完整,极少破碎。随着ATTM投料质量分数的增加,颗粒表面不再如模板光滑,表面逐渐出现大量沟壑。当ATTM投料60%时MoS2/C-60-SBA-15结构坍塌(图2(g)和2(h))。
图3为SBA-15、MoS2/C-1-SBA-15和MoS2/C-10-SBA-15的透射电镜照片(TEM)。图3(b)可以看到SBA-15高度有序的介孔孔道。图3(c)和3(e)看到MoS2/C-1-SBA-15和MoS2/C-10-SBA-15具有与SBA-15相同的形状,且孔道结构与SBA-15相似(图3(d)和3(f)),高倍透射电镜(HRTEM) (图3(g))显示,MoS2/C-10-SBA-15中的碳主要为无定形炭,少量的少层MoS2片分散在无定形炭中。MoS2/C-40-SBA-15(图4(a))的颗粒完整,整体形貌与模板SBA-15相似,可以看到有序介孔的结构。图4(b)为MoS2/C-40-SBA-15通过超声破碎机处理后的TEM照片,可以更清楚地看到孔道和柱状的孔壁,其中,少层MoS2均匀嵌入无定形炭中共同组成了有序介孔材料的骨架。图4(c)中可以观察到少层(2-4层)的MoS2均匀分布在无定形炭中,测量MoS2的层间距为0.65 nm,与XRD谱图中14.7°处出现的MoS2(002)晶面特征峰相匹配。此外,能量色散X射线光谱(EDS)(图4(d))可以表明C、S、Mo元素均匀分布于介孔材料中。图4(e)为未破碎MoS2/C-60-SBA-15的TEM照片,可以看到样品颗粒中有黑色的团聚物,存在分布不均匀的情况。HRTEM照片4(f)显示 MoS2的层数为10-15层,层数较多不再具有少层的特性,这归因于MoS2量过多,无定形炭无法很好分散MoS2,从而出现了多层MoS2团聚的现象。
图5为SBA-15和MoS2/C-X-SBA-15的小角度(图5(a))及大角度(图5(b))X射线衍射(XRD)谱图。可以清楚地看到SBA-15硬模板在小角度范围存在三个衍射峰,分别出现在0.97°、1.63°和1.87°处,分别对应p6mm对称六边形布拉格反射的(100)、(110)和(200)[27]。图5(a)中可以发现,MoS2/C-1-SBA-15同样存在三个衍射峰,这说明制备的MoS2/C-1-SBA-15具有有序介孔结构,但相较于SBA-15存在明显右移。根据布拉格衍射公式2dsinθ=nλ,θ值增大,则d值减小,通过公式(a0=)估算得到晶胞参数(a0)(表1),发现MoS2/C-1-SBA-15的晶胞参数小于模板SBA-15的晶胞参数,这是由于高温煅烧使铸造样品的尺寸相较于SBA-15模板存在一定收缩[24]。图5(b)中,MoS2/C-1-SBA-15在22°附近存在宽峰,对应样品中存在无定形炭。MoS2/C-(10-40)-SBA-15在图5(a)中可以观察到在1.05°处有明显的衍射峰,在1.80°和2.00°处有两个峰强度较弱的衍射峰,这与MoS2/C-1-SBA-15的衍射峰一致,说明MoS2/C-(1-40)-SBA-15具有有序的介孔结构。MoS2/C-(10-60)-SBA-15在图5(b)中可以观察到在14.4°、32.7°、39.5°和58.3°处存在4个明显的特征衍射峰,这可以很好对应六边晶系2H-MoS2的(002)、(100)、(103)和(110)晶面,与JCPDS:37-1492卡片相一致。MoS2/C-1-SBA-15在图5(b)中仅观察到14.4°处存在衍射峰,与2H-MoS2的(002)晶面相对应,这是由于ATTM投料仅为1%,煅烧后生成的MoS2较少,衍射峰不明显;在22°处观察到明显宽峰,说明材料中存在无定形炭。随着ATTM投料质量分数的增加,MoS2对应衍射峰强度增强,无定形炭对应的宽峰减弱。当ATTM投料量达到60%时MoS2/C-60-SBA-15在图5(a)中无特征衍射峰,说明样品存在有序介孔坍塌的情况,导致高度有序介孔结构消失。
图6为硬模板SBA-15和MoS2/C-X-SBA-15材料的氮气吸附-脱附曲线。由图6中可以看出,SBA-15展示出了典型的IV型曲线,具有H1型回滞环,表明SBA-15具有标准的圆柱形中孔通道[27],孔隙体积为1.12 cm3/g。MoS2/C-(1-40)-SBA-15的吸附等温线与介孔碳的曲线相似,为IV型吸附等温线[29],表明材料具有介孔结构。MoS2/C-(1-40)-SBA-15复合材料的孔尺寸反应了硬模板的孔壁厚度。随着ATTM投料比例的增加,MoS2逐渐成为支撑介孔复合材料的主要成分,当ATTM投料达到40%时,MoS2/C-40-SBA-15吸附等温曲线在0.9-1.0时,吸附量迅速增加,表明存在一些结构孔隙[24],这可能是因为在管式炉煅烧过程中ATTM分解生成MoS2和H2S造成了体积损失,从而使MoS2/C不能完全填充SBA-15的孔道。当ATTM投料达到60%时,MoS2/C-60-SBA-15的回滞环几乎不可见。由图6(b)可以发现,MoS2/C-(1-40)-SBA-15的孔径尺寸相近,孔径较为均匀。但MoS2/C-60-SBA-15的孔径分布较宽,说明孔径不均匀。MoS2/C-(1-60)-SBA-15的比表面积随着ATTM投料增加而下降(表1),当ATTM投料40%时MoS2/C-40-SBA-15的比表面积为348 m2/g,比表面积仍然较高,当ATTM投料达到60%时MoS2/C-60-SBA-15的比表面积仅为72 m2/g,比表面积明显降低,表明MoS2/C-60-SBA-15样品孔道坍塌、结构破碎的情况较多,与XRD小角度无特征峰结果相一致。
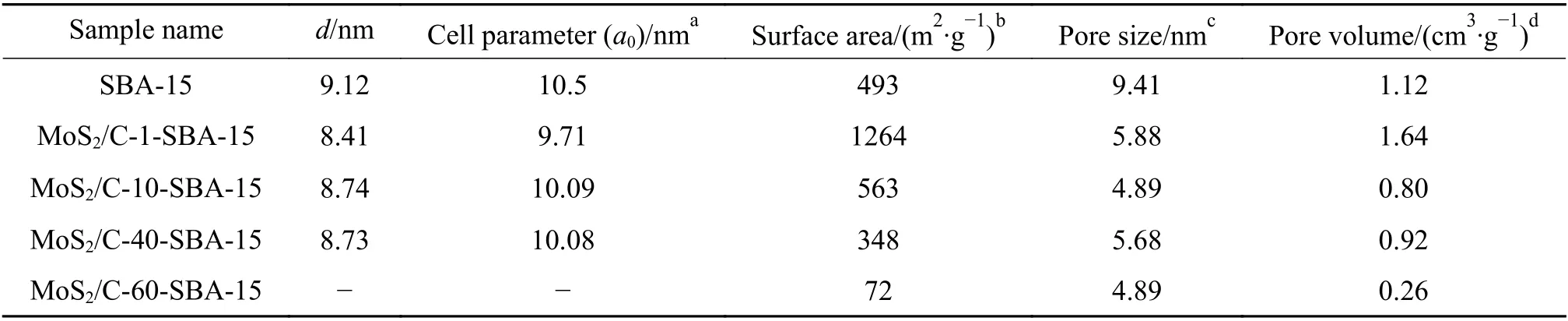
表1 样品的介孔结构Table 1 Textural Properties of Mesostructured Products
图7为MoS2/C-X-SBA-15的Raman光谱图。从图7(a)中可以看出,MoS2/C-X-SBA-15在376和402 cm-1处两个波段存在两个峰,分别对应2HMoS2的和A1g两种模式[30]。该结果与XRD的结论相一致。 图7(b)中可以看到,在1342与1587 cm-1处存在分别对应无序碳结构的D峰与对应sp2碳原子的G峰。通过计算ID/IG可以衡量碳材料石墨化程度的高低[31],碳的石墨化有利于电化学反应过程中电子的运输转移。分析计算结果发现样品ID/IG值相近,说明MoS2的引入没有影响样品中碳的石墨化程度。
X射线光电子能谱(XPS)测量揭示了MoS2/C-40-SBA-15的表面化学状态。XPS测量显示MoS2/C-40-SBA-15中存在Mo、S、C、O四种元素(图8(a))。在Mo 3d的高分辨率光谱中(图8(c)),Mo 3d5/2和Mo 3d3/2分别在结合能为229.6和232.7 eV处出现一对峰,对应为Mo4+物种[32],是MoS2样品中Mo的主要存在形式。在233.3和236.4 eV处出现了一对与Mo6+对应的Mo 3d3/2和Mo 3d5/2峰,这可能是由于样品中O对周围MoS2组分的电荷诱导效应,使少量Mo拥有更高的结合能[33]。此外,在226.9 eV处还出现了与S 2s(MoS2)相关的峰[33]。在S 2p区域(图8(d)),样品在结合能为162.5和163.6 eV处出现了两个与MoS2有关的峰,分别对应S 2p3/2和S 2p1/2[34]。在163.9和165.1 eV出现的两个峰对应,表示在S-Mo-S层的顶部和底部存在不饱和的S原子,即边缘S位点[35-37]。同时,在168.9 eV处也观察到一个峰,表明在最终的样品中存在S6+-O(如的形式)[33]。在C 1s谱图(图8(b))中,可以看到在284.8 eV处存在一个强峰,对应样品中的C-C,而在结合能为285.9和290.1 eV处存在两个弱峰,分别对应C-O-C和O-C=O[35,38]。样品中少量O可能来源于样品表面在空气中氧化。
2.2 MoS2/C-X-SBA-15复合材料催化性能
对MoS2/C-X-SBA-15进行多种电化学性能测试(图9)。采用线性扫描伏安法(LSV)对材料测试(图9(a)),当电流密度为10 mA/cm2时,MoS2/C-(1-60)-SBA-15过电位分别为355、236、165和170 mV。通过计算Tafel斜率来衡量材料的反应动力学活性(图9(b)),显示MoS2/C-40-SBA-15塔菲尔斜率最小,为91.1 mV/dec,表示该比例下HER更迅速。EIS图显示MoS2/C-X-SBA-15样品阻抗特性相似,表明它们的电化学过程相似,建立等效模拟电路图,其中,Rs为电解质电阻、Rct为电荷转移电阻和CPE恒相元件(图9(c))。MoS2/C-(1-60)-SBA-15的电荷转移电阻(Rct)值分别为18.24、6.93、3.86和4.56 Ω,MoS2/C-40-SBA-15的Rct值最小,说明MoS2/C-40-SBA-15比其他材料具有更好的电荷转移性能。MoS2/C-40-SBA-15是在可以保持三维有序介孔结构前提下MoS2含量最高的样品,说明具有三维结构并实现少层MoS2均匀分散的样品,相较于形貌为块体且出现MoS2团聚的样品,具有更高的电荷转移能力。稳定性也是衡量电化学催化剂的重要指标(图9(d)),对MoS2/C-40-SBA-15样品施加10 mA/cm2的电流,测量其电压的变化,持续测量24 h,发现其具有较好的稳定性能。通过SEM对电化学稳定性测试后的样品进行形貌分析(图10(a)、(b)),可以看出MoS2/C-40-SBA-15样品颗粒的整体尺寸和形貌没有改变与反应前的形貌一致,结合前文的表征可以说明催化剂在稳定性测试后保持了原有的三维有序介孔结构。采用双层电容法,根据ECSA=Cdl/Cs测量电化学活性比表面积(ECSA)[39](图11),在0.04-0.14 V(vs.RHE)电压下,以不同扫速对材料进行循环伏安测试,结果如图11(e),MoS2/C-40-SBA-15的Cdl值最大(57.9 mF/cm2),说明MoS2/C-40-SBA-15拥有最大的活性面积。这是由于三维有序的介孔结构可以为反应提供较大的比表面积,被均匀分散的少层MoS2片可以提供大量的析氢活性位点。
结合材料结构表征和电化学析氢性能分析,MoS2为MoS2/C-X-SBA-15复合材料的主要析氢活性组分,但MoS2的含量并不是决定性能的唯一参数,催化剂的三维结构和MoS2片层的分布,对电化学析氢性能都有至关重要的影响。
3 结 论
本研究以SBA-15为硬模板,蔗糖为碳源,受热原位解为MoS2的四硫代钼酸铵为前驱体,通过纳米铸造牺牲硬模板的方法合成了具有三维有序介孔结构的MoS2/C-SBA-15复合材料。整个制备过程方便、环保,没有释放大量的H2S有害气体。制得催化剂中少层(2-4层)MoS2片均匀的分散在介孔材料的碳骨架中,使MoS2片边缘的活性位点得以暴露在高比表面积介孔材料的表面。当ATTM投料为40%时制备了MoS2/C-40-SBA-15,比表面积高达348 m2/g,在电流密度为 10 mA/cm2时,过电位为165 mV,Tafel 斜率约为91.1 mV/dec。MoS2/C构建的三维有序介孔析氢催化剂,不仅提供了大量活性位点,还为反应过程中的物质交换提供了传输通道。MoS2薄片均匀分散在三维碳骨架中相较于MoS2分散不均、结构破碎的块体具有更小的阻抗,有利于电子快速转移。综上,本实验为制备结构可控、活性组分均匀分散的三维结构电化学析氢催化剂提供了一种可行的方法。
猜你喜欢
现代制造技术与装备(2022年4期)2022-05-28
粮食加工(2022年1期)2022-03-23
玻璃(2022年1期)2022-02-23
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
燃料化学学报(2019年10期)2019-11-04
表面工程与再制造(2019年6期)2019-08-24
汽车文摘(2018年1期)2018-11-26
分析化学(2018年4期)2018-11-02
广东教育·高中(2018年12期)2018-02-13
