
NADPH氧化酶4调控组蛋白脱乙酰酶4在内皮素1诱导的体外大鼠心肌细胞肥大中的作用*
2022-12-03 08:46王燕鸽赵昕张蕾古心雨雷婵豪李瑞芳
中国病理生理杂志 2022年11期
王燕鸽,赵昕,张蕾,古心雨,雷婵豪,李瑞芳
(河南科技大学基础医学院药学系,河南 洛阳 471023)
血管烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate,NADPH)氧化酶(NADPH oxidase,NOX)激活引起的活性氧(reactive oxygen species,ROS)生成增多在心肌肥大的发病过程中发挥关键作用[1],但是在心肌肥大发病中何种NOX亚型是有害的因素,何种NOX亚型是保护的因素尚不明确,NOX调控的下游信号通路还有待进一步研究[2]。前期研究显示肾性高血压大鼠心脏中NOX4的表达随着心肌肥大程度增加呈上升趋势,并且随着心肌肥大的发展组蛋白脱乙酰酶(histone deacetylase,HDAC)的表达也发生了变化,提示NOX4与HDAC均参与了高血压左心室肥大的发病过程[3-4]。探讨NOX4在心肌肥大中的调节模式,进一步揭示NOX4对肥大信号的融合节点HDAC4和心肌肥大负性调控因子糖原合成酶激酶3β(glycogen synthase kinase-3β,GSK-3β)的调控机制,对研发心血管疾病的新靶点药物具有重要价值。
材料和方法
1 动物
SPF级健康雌性SD大鼠,由河南科技大学实验动物中心提供,许可证号为SCXK(豫)2019-0002。实验使用2~3日龄新生SD乳大鼠,分离培养原代心肌细胞。
2 主要试剂
内皮素1(endothelin-1,ET-1)购自Sigma;NOX4抑制剂GKT137831购自Selleck;ECL化学发光检测试剂盒和BCA蛋白浓度测定试剂盒购自Pierce;RNase抑制剂购自北京索莱宝科技有限公司;Trizol核酸提取试剂购自Invitrogen;二氢乙啶(dihydroethidium,DHE)购自上海碧云天技术有限公司;抗NOX4抗体、抗p-Akt抗体、抗p-GSK-3β抗体和抗p-HDAC4抗体(Abcam);抗p47phox抗体和抗α-tubulin抗体(Proteintech)。实验引物由上海工生物工程股份有限公司设计合成。
3 方法
3.1 原代心肌细胞的分离与培养取2~3日龄的SD乳鼠进行原代心肌细胞分离培养。按照本实验室建立改良的心肌细胞分离培养的方法进行[5],乳鼠心肌组织用0.05 g/L DNase I和0.1%胰蛋白酶消化成单细胞悬液。差速贴壁分离后,加入含10%胎牛血清和0.1 mmol/L BrdU的DMEM培养液,在37℃、5%CO2培养箱中培养24 h后,更换培养液,进行后续实验。
3.2 实验分组将心肌细胞以每孔1×108/L的浓度接种于6孔板中,加入含10%胎牛血清和0.1 mmol/L BrdU的DMEM培养液,细胞于24 h贴壁后,用无菌PBS冲洗2~3遍清洗死细胞,加入含10%胎牛血清DMEM培养液24 h,不同试剂对其进行处理,并分为以下4组:(1)空白对照(control)组:以含10%血清的DMEM培养液培养24 h;(2)GKT137831干预组:20µmol/L GKT137831处理24 h[6];(3)ET-1造模组:0.1µmol/L ET-1处理24 h[5];(4)GKT137831+ET-1干预组:20µmol/L GKT137831加0.1µmol/L ET-1处理24 h。
3.3 ROS检测将1.5 h差速贴壁后的原代心肌细胞接种于6孔板中,37℃、5%CO2下的潮湿细胞培养箱中培养24 h至细胞贴壁。心肌细胞24 h贴壁后分组处理,再于37℃、5% CO2下的潮湿细胞培养箱中培养24 h,显微镜下观察细胞状态。用无菌PBS冲洗2~3遍,然后加入DHE染色试剂,使终浓度为5µmol/L,并在37℃、5% CO2下远离光线的环境中一起孵育30 min。然后弃去培养液,PBS清洗细胞3次,荧光显微镜观察并采集图像。
3.4 Western blot采用RIPA裂解液提取心肌细胞总蛋白。使用BCA蛋白质检测试剂盒检测蛋白质浓度,并将上样量调整至50µg。通过10% SDS-PAGE分离蛋白,然后转移至PVDF膜,5%脱脂奶粉室温封闭1 h,加入Ⅰ抗于4℃孵育过夜,与辣根过氧化物酶(HRP)标记的Ⅱ抗室温孵育1 h。ECL化学发光显色法检测蛋白条带。用ImageJ软件分析蛋白质条带的灰度值。
3.5 RT-qPCR采用Trizol提取各组细胞的总RNA,用RNA纯度分析仪检测RNA浓度。参照逆转录试剂盒操作说明进行逆转录,总反应体积20µL:2×RT Mix 10µL,HiScript II Enzyme Mix 2µL,Oligo(dT)23VN(50µmol/L)1µL,random hexamers(50 mg/L)1µL,RNA 1µg。反应条件为:25℃5 min,50℃15 min,85℃2 min。参照RT-qPCR试剂盒操作说明进行RT-qPCR,总反应体积20µL:2×AceQ qPCR SYBR Green Master Mix 10.0µL,0.4µL 50×ROX Reference Dye 2,1µL DNA,0.4µL引物。扩增条件为:95℃预变性5 min;95℃变性10 s,60℃1 min,95℃15 s,40个循环;60℃1 min,95℃15 s。然后,通过RT-qPCR扩增产物的熔解曲线分析数据,分析所测基因的Ct值和特异性,以18S为内参照,分析ET-1诱导的心肌细胞中肥大标志物心房钠尿因子(atrial natriuretic factor,ANF)和β-肌球蛋白重链(β-myosin heavy chain,β-MHC)的mRNA表达水平。引物序列见表1。

表1 RT-qPCR物序列Table 1.Sequences of the primers for RT-qPCR
4 统计学处理
实验所得数据以均数±标准差(mean±SD)表示,应用SPSS 24.0软件进行统计分析,使用GraphPad Prism 7进行柱状图绘制。组间均数比较采用单因素方差分析,两两比较采用LSD-t检验。以P<0.05为差异有统计学意义。
结 果
1 抑制NOX4对ET-1诱导的心肌细胞表面积及ANF和β-MHC mRNA表达的影响
心肌肥大的特征性表型变化包括心肌细胞表面积的增大,以及肥大标志物ANF和β-MHC的表达。因此,我们观察了GKT137831对ET-1诱导的心肌细胞表面积的影响。结果显示,与空白对照组相比,ET-1刺激使心肌细胞表面积显著增大;GKT137831处理可显著减轻由ET-1引起的心肌细胞表面积的增加(P<0.01),见图1A。RT-qPCR检测GKT137831对ET-1诱导的心肌细胞肥大标志物ANF和β-MHC表达的影响,结果显示,与空白对照组相比,ET-1刺激显著提高了心肌细胞中ANF和β-MHC的mRNA表达水平,而GKT137831处理则显著抑制了ET-1诱导的心肌细胞中ANF和β-MHC的mRNA表达(P<0.01),见图1B。
2 抑制NOX4对ET-1诱导的心肌细胞中ROS生成的影响
采用DHE染色法检测GKT137831对ET-1诱导的心肌细胞中ROS生成的影响。结果显示,与对照组相比,ET-1刺激心肌细胞肥大后,显著增加了心肌细胞中ROS的生成(P<0.05),而GKT137831显著减少了ET-1诱导的肥大心肌细胞中ROS的产生(P<0.01),见图2。
3 抑制NOX4对ET-1诱导的心肌细胞中NOX表达的影响
采用Western blot法 检测GKT137831对ET-1诱导的心肌细胞中NOX表达的影响。结果显示,与空白对照组相比,经过ET-1诱导后使肥大心肌细胞中NOX催化亚基NOX4和调节亚基p47phox的蛋白表达显著升高(P<0.05或P<0.01),而GKT137831显著抑制了ET-1诱导的肥大心肌细胞中NOX4和p47phox的蛋白表达(P<0.01),见图3。
4 抑制NOX4对ET-1诱导的心肌细胞中Akt/GSK-3β通路的影响
为了研究抑制NOX4对ET-1诱导的心肌肥大的潜在机制以及ET-1和GKT137831对Akt/GSK-3β通路的影响,我们采用Western blot法检测GKT137831对ET-1诱导的心肌细胞中磷酸化Akt/GSK-3β蛋白水平的影响。结果显示,与空白对照组相比,经过ET-1刺激后肥大心肌细胞中Akt和GSK-3β的磷酸化水平显著升高(P<0.05或P<0.01);而GKT137831处理显著抑制了ET-1诱导的Akt和GSK-3β的磷酸化(P<0.01),见图4。
5 抑制NOX4对ET-1诱导的心肌细胞中HDAC4磷酸化的影响
采用Western blot法 检测GKT137831对ET-1诱导的心肌细胞中HDAC4磷酸化水平的影响,结果显示,与空白对照组相比,由ET-1刺激的肥大心肌细胞经过GKT137831处理后,GKT137831显著降低了ET-1诱导的心肌细胞中HDAC4的磷酸化水平(P<0.01),见图5。
讨 论
心力衰竭是多种心脏疾病的终末阶段,是全球住院和死亡的主要原因之一,长期存在的心肌肥大可以导致心功能障碍和心力衰竭[7]。ET-1是一种血管收缩剂,可以刺激内皮素系统和肾素-血管紧张素-醛固酮系统,促进心脏收缩,诱导心肌肥大[8]。在本研究中,通过在体外使用ET-1诱导的心肌肥大模型,我们检测到NOX4是心肌肥大的正向调节因子,抑制NOX4可以防止ET-1诱导的心肌细胞中Akt/GSK-3β-HDAC4信号通路的激活,进而防止和逆转心肌肥大。

Figure 1.Effects of GKT137831 on the surface area(A)and the ANF andβ-MHC mRNA levels(B)in ET-1-induced cardiomyocytes.The scale bar=50µm.Mean±SD.n=3.**P<0.01 vs control group;##P<0.01 vs ET-1 group.图1 GKT137831对ET-1诱导的心肌细胞表面积及肥大标志物ANF和β-MHC mRNA表达的影响
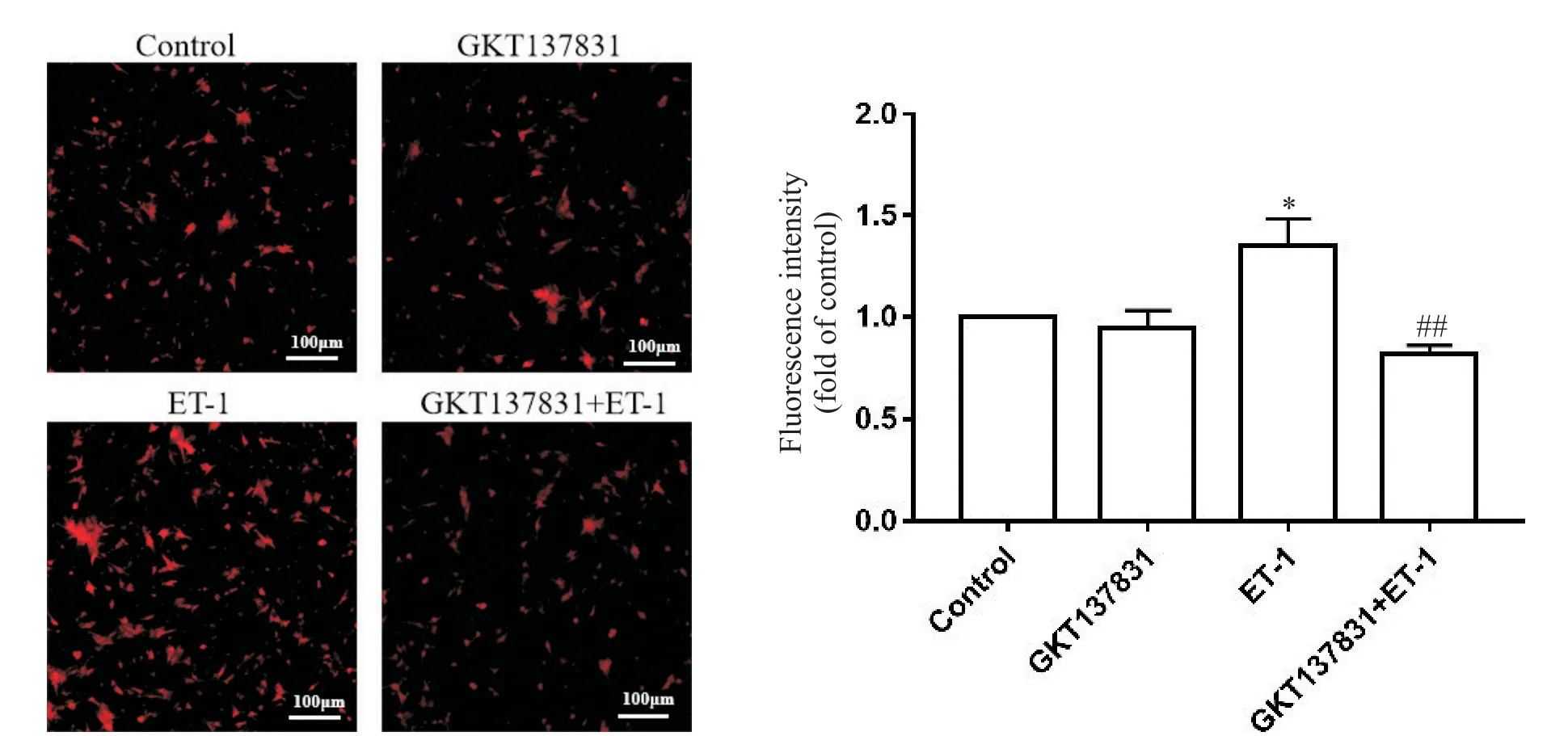
Figure 2.Effects of GKT137831 on ROS generation in ET-1-induced cardiomyocytes.The scale bar=100µm.Mean±SD.n=3.*P<0.05 vs control group;##P<0.01 vs ET-1 group.图2 GKT137831对ET-1诱导的心肌细胞ROS生成的影响

Figure 3.Effects of GKT137831 on the protein expression of NOX4 and p47phox in ET-1-stimulated cardiomyocytes.Mean±SD.n=3.*P<0.05,**P<0.01 vs control group;##P<0.01 vs ET-1 group.图3 GKT137831对ET-1刺激心肌细胞中NOX4和p47phox表达的影响
NOX的催化亚基在心血管细胞中包含多个亚型,它们在不同病理刺激因素作用下介导不同的病理过程,其作用机制尚不明确[9]。NOX4基因敲除可以显著减轻动脉缩窄诱导的小鼠心肌肥大,改善心脏功能,减少ROS生成[10];但是,也有不同的报道显示NOX4基因敲除小鼠在慢性压力负荷诱导下可以出现严重的扩张性心肌肥大和心脏收缩功能失调,而NOX4转基因小鼠则受到保护[11]。研究者认为这种矛盾的产生主要是由于基因敲除的程度和手术的方法不同,但这种解释没有足够的说服力,关于NOX各亚型在不同病理发展过程中的作用尚需大量的实验来确证。因此,在本研究采用NOX4抑制剂GKT137831抑制肥大心肌细胞中NOX4的表达,结果观察到其可以降低ET-1诱导的NOX4和p47phox的蛋白表达和ROS的生成,对ET-1刺激造成的氧化应激和病理性心肌肥大有一定的治疗作用,进一步证明NOX4参与心肌肥大。
在小鼠心脏特异性过表达构成性激活的GSK-3β,可以抑制动脉缩窄或异丙肾上腺素刺激诱导的心肌肥大,也可以部分防止激活的钙调磷酸酶引起的心肌肥大反应。ET-1和压力超负荷都可能通过PI3K-Akt通路抑制GSK-3β,导致心肌肥大发生。这些研究表明GSK-3β在调节心肌肥大中起了至关重要的作用。GSK-3β可以磷酸化一系列的转录因子(NFAT、GATA4、c-Jun、c-Myc和cyclin D1),从而发挥抗心肌肥大作用[12]。在心肌肥大中NOX4是否通过调控GSK-3β,进而影响与肥大相关的转录因子的表达,是研究中要解决的另一个问题。因此,本研究抑制肥大心肌细胞中NOX4的表达,结果显示GKT137831减弱了ET-1诱导的心肌细胞表面积增加、肥大标志物ANF和β-MHC的mRNA表达以及Akt和GSK-3β的磷酸化,表明抑制NOX4减轻了ET-1诱导的心肌细胞肥大,该作用与其抑制肥大信号通路Akt/GSK-3β的激活有关。
在心肌肥大十分复杂的信号网络中,HDAC是一个重要的下游融合节点,调节许多与肥大相关的转录因子表达,可能是治疗心肌肥大的一个很有意义和前景的作用靶标[13]。目前,国外关于HDAC的研究主要集中在抗肿瘤领域,已经有一部分HDAC抑制剂开始用于临床恶性肿瘤的治疗[14];而在心肌肥大发病中的研究,主要是通过基因敲除小鼠来阐明不同HDAC亚型的功能[15]。迄今为止,HDAC在高血压心肌肥大中的作用及以其为靶标的药物治疗,国内外还未见报道。在本研究中,我们检测了由ET-1诱导的肥大心肌细胞中HDAC4蛋白的磷酸化水平,显示GKT137831可以显著降低HDAC4蛋白的磷酸化。因此,GKT137831抑制ET-1诱导的心肌肥大反应,该作用与HDAC4有关。NOX4可以影响ET-1诱导的心肌细胞中HDAC4的磷酸化水平。

Figure 4.Effects of GKT137831 on the phosphorylation of Akt and GSK-3βin ET-1-stimulated cardiomyocytes.Mean±SD.n=3.*P<0.05,**P<0.01 vs control group;##P<0.01 vs ET-1 group.图4 GKT137831对ET-1刺激心肌细胞中Akt和GSK-3β磷酸化的影响

Figure 5.Effect of GKT137831 on the phosphorylaton of HDAC4 in ET-1-stimulated cardiomyocytes.Mean±SD.n=3.*P<0.05 vs control group;##P<0.01 vs ET-1 group.图5 GKT137831对ET-1刺激心肌细胞中HDAC4磷酸化的影响
综上所述,本研究表明抑制NOX4显著减轻了ET-1诱导的心肌细胞肥大,减少了NOX的表达和ROS的生成,防止了Akt/GSK-3β-HDAC4信号通路的激活,部分逆转心肌肥大。该研究为NOX4在心肌肥大发病中的调节机制提供实验依据。
猜你喜欢
小学生学习指导(高年级)(2022年3期)2022-03-29
波谱学杂志(2022年1期)2022-03-15
中学生物学(2021年8期)2021-11-02
天津医科大学学报(2019年6期)2019-08-13
中国果业信息(2019年1期)2019-01-05
中国畜牧兽医文摘(2018年6期)2018-07-28
分析化学(2017年12期)2017-12-25
小学生导刊(高年级)(2017年2期)2017-06-10
小学生导刊(2017年6期)2017-02-10
小学生导刊(高年级)(2016年1期)2016-01-29
