
一种面向药物-靶点相互作用预测的不平衡数据处理方法
2022-11-30 08:03叶志威张晓龙林晓丽
武汉科技大学学报 2022年1期
叶志威,张晓龙,林晓丽
(1.武汉科技大学计算机科学与技术学院,湖北 武汉,430065;2.武汉科技大学大数据科学与工程研究院,湖北 武汉,430065;3.武汉科技大学智能信息处理与实时工业系统湖北省重点实验室,湖北 武汉,430065)
Word2Vec模型非常适合于处理类似药物分子和蛋白质序列信息这样的原始文本数据。它将原始数据当成文章,学习其中每个分词之间的联系(分词为序列中一个一个的小片段,可以理解为药物分子和蛋白质靶点的子结构),这种联系包含了丰富的药物-靶点相互作用信息,将其以实数向量的形式表示出来,可以使原始的非数值数据数值化,从而方便更深入的算法研究。
在过往的生物学研究中,只有可以相互作用的数据(正样本)被当成有意义的信息而收集记录下来,因此在药物-靶点相互作用研究中,数据集往往是不平衡的,即缺少负样本,同时未知的药物-靶点相互作用数据又数倍于已知的药物-靶点相互作用数据。针对这一问题,Luo等[11]通过随机抽样来提取负样本子集,但这些负样本数据集中也可能存在药物-靶点相互作用的正样例。Peng等[12]则通过PU-learning这种半监督学习方法来解决这个问题。在此基础上,Lin等[13]提出一种基于非随机缺失标签的模型,取得了更好的结果。另外,Chawla等[14]提出SMOTE算法,根据少数类样本合成新样本并添加到数据集中。Han等[15]在SMOTE基础上进行了改进,提出Borderline-SMOTE算法,仅使用边界上的少数类样本来合成新样本,从而改善了样本的类别分布。
本文提出一种面向药物-靶点相互作用预测的不平衡数据处理方法BS-DTVec。首先基于Word2Vec模型,分别获取药物分子的低维特征表示(DrugVec)和靶点蛋白质的低维特征表示(TargetVec),并将它们组合为药物-靶点相互作用对的低维向量特征表示(DTVec);然后通过实验模型从原始的药物和蛋白质序列数据中学习药物-靶点相互作用的关键信息。在此过程中,针对数据集的不平衡性可能引起的过拟合问题,采用Borderline-SMOTE技术合成少数类样本;最后以梯度提升决策树(Gradient Boosting Decision Tree, GBDT)作为分类器对预处理后的数据进行分类预测。
1 BS-DTVec方法设计
1.1 数据集
本研究主要通过KEGG数据库[16]和DrugBank数据库[17]中的数据进行药物-靶点相互作用预测。4种蛋白质靶点已经被Yamanishi等[1]用作黄金标准数据集,它们分别是酶(Enzyme)、离子通道(Ion Chanel)、G蛋白偶联受体(GPCR)和核受体(Nuclear Receptor)。黄金标准数据集可以从http://web.kuicr.kyoto-u.ac.jp/supp/yoshi/drugtarget/网址获取。另外还选取了来自DrugBank 4.3的数据,该数据可以在DrugBank官网https://go.drugbank.com/上获取。黄金标准数据集和DrugBank 4.3数据集的详细信息如表1所示。
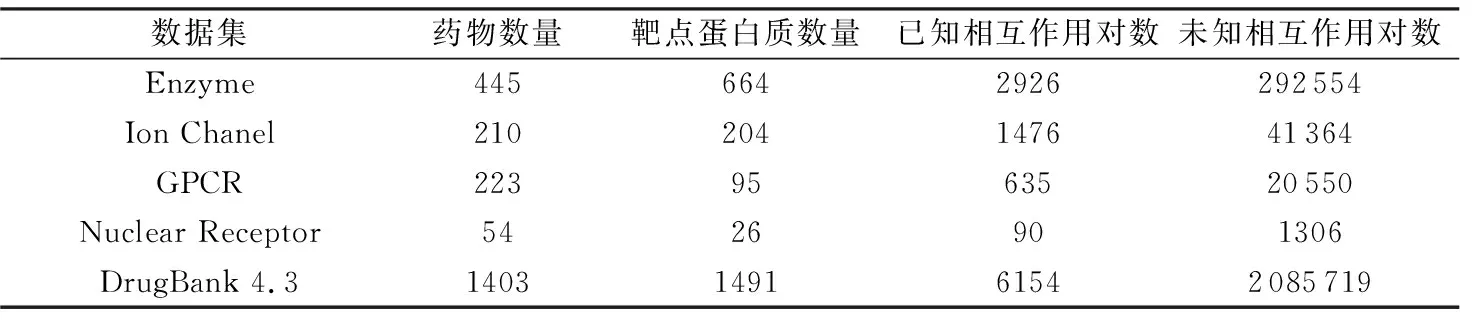
表1 黄金标准数据集和DrugBank 4.3数据集信息
1.2 少数类样本合成
面临数据集不平衡问题时,随机过采样是最简单、最直接的处理方法。该方法通过直接复制少数类样本来平衡数据集,但是这种方法容易产生过拟合的问题,使得模型学习到少数类样本的部分非主要特征,即模型学习到的特征过于特别而不具备普适性。SMOTE算法不再是通过简单的复制来生成样本,而是通过对少数类样本进行分析后重新合成新样本并添加到原始数据集中。这种新样本是由已知的少数类样本分析得到的,因此具有少数类样本的通性但又不同于原始少数类样本。在样本空间中,SMOTE生成新样本的公式如下:
xnew=x+rand(0,1)·(x′-x)
(1)
式中:x′为少数类样本x的近邻样本;xnew为生成的新样本。
首先根据少数类样本和多数类样本的比例自主设定采样倍率N。对于每一个少数类样本x,找到K个和x最相近的少数类样本,然后从中随机选取N个邻近样本,按式(1)随机生成新样本xnew,这样就将少数类样本增加了N倍。SMOTE算法在生成新样本时容易产生数据分布边缘化的问题。如果一个少数类样本处在样本空间的边界位置,这个样本与其邻近样本生成的新样本也会在边界位置。大量边界样本的生成会模糊原少数类样本与多数类样本的边界,从而加大了对数据集分类的难度。
本文采用的Borderline-SMOTE算法是对SMOTE算法的改进。假设原始训练样本集为T,其中少数类为X、多数类为Y,并且X={x1,x2,…,xi,…,xxnum},Y={y1,y2,…,yi,…,yynum},xnum、ynum分别为少数类样本个数和多数类样本个数。与SMOTE相比,Borderline-SMOTE算法只针对边界样本进行近邻线性插值,使得合成后的样本分布更加合理。Borderline-SMOTE算法描述如下。
高等学校担负着为祖国输送高素质人才的光荣使命,心理健康教育在高等学校人才培养工作中发挥着不可忽视的作用。而高素质人才首先要有一个积极向上的心态,换而言之就是心理健康对高素质人才有着决定性的作用。

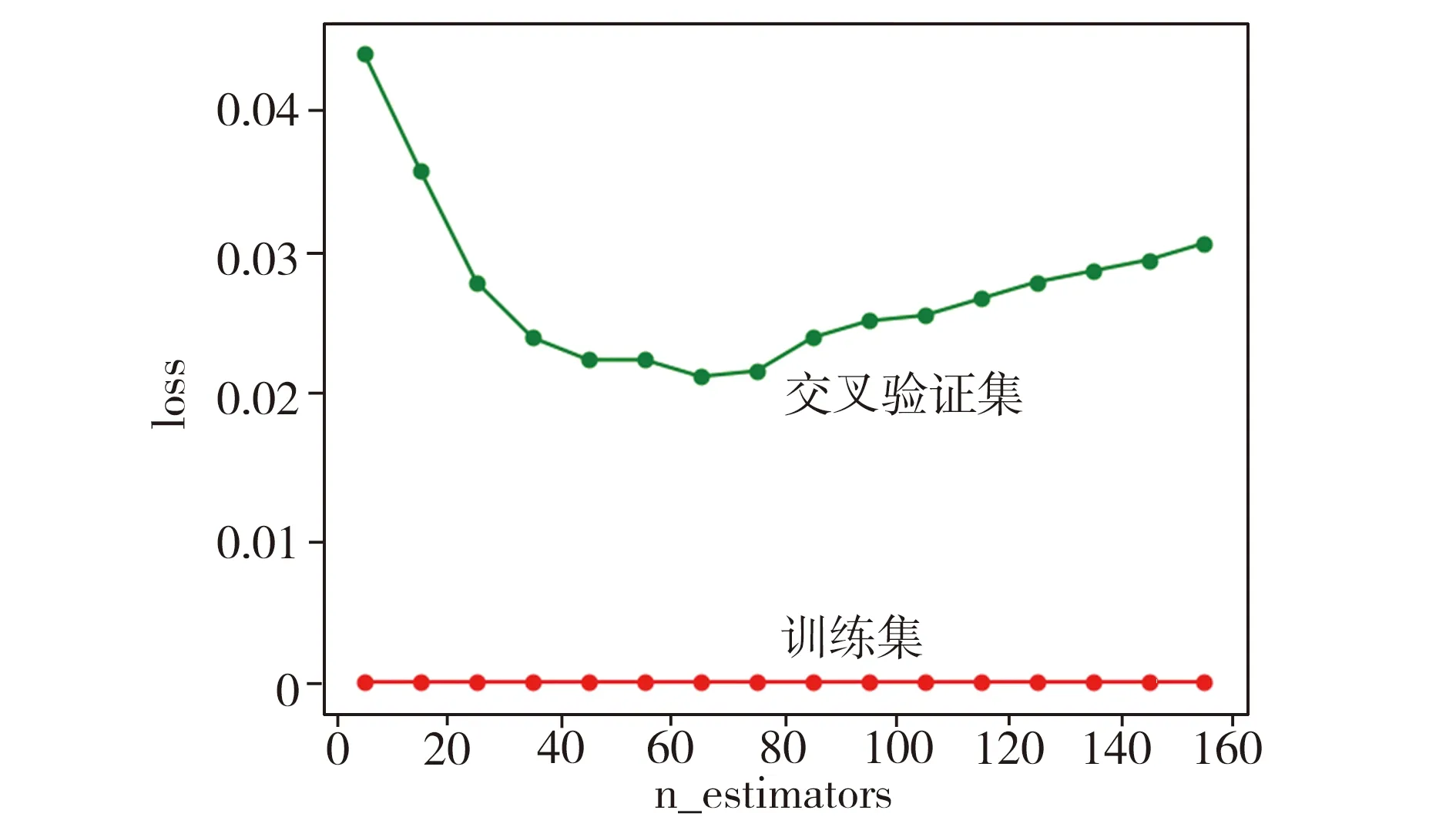
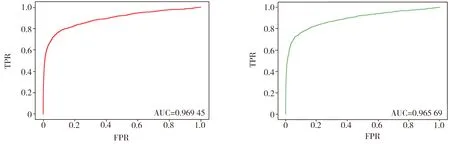
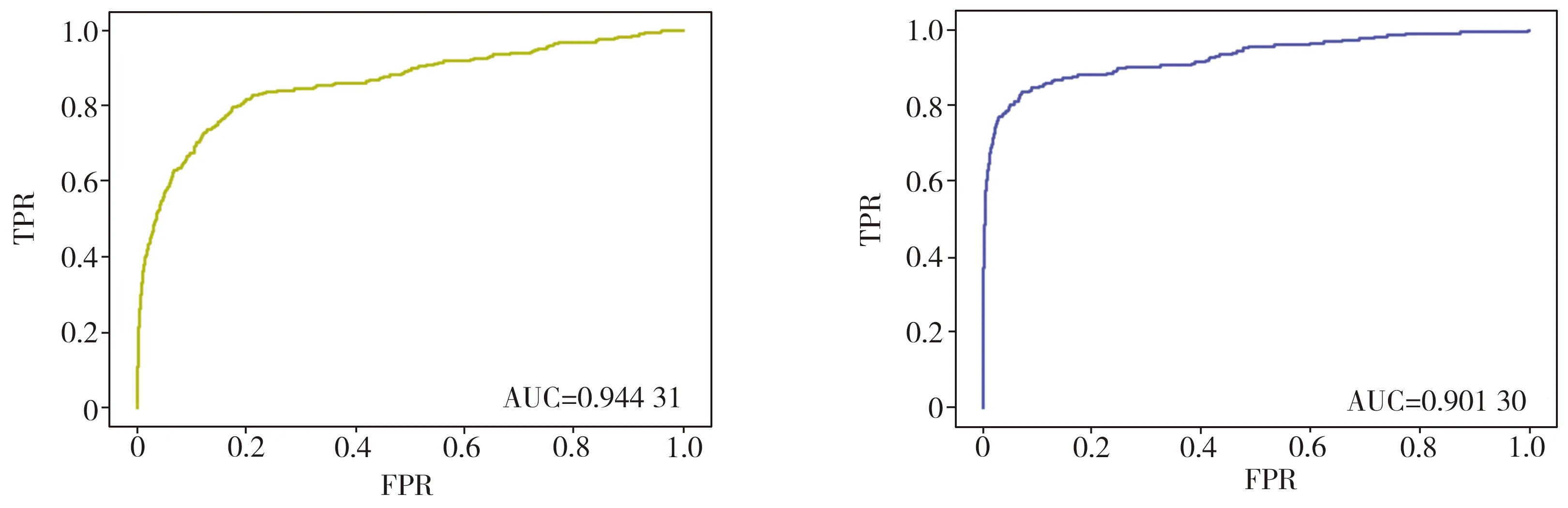
输入:药物-靶点组合特征和标签组成的数据集T,采样倍率N。 输出:指定正负比例的数据集T'。 第一步:计算少数类样本中每个样本点xi与所有其他训练样本的欧氏距离,获取该样本的K近邻。 第二步:对少数类样本进行划分,设K近邻中有K'个属于多数类样本(0≤K'≤K): 如果K'=K,则xi被认为是噪声; 如果K2≤K' 本文提出的DTVec是受Word2Vec启发的药物分子和蛋白质靶点的特征表示方法。Word2Vec模型通过训练可以将语句中的每个词映射成一个低维实数向量。常见的Word2Vec模型有CBOW和Skip-gram两类,本文选用Skip-gram模型。Skip-gram模型采用一个三层(输入层-隐藏层-输出层)的神经网络,整个模型的关键是根据词语出现的频率实现Huffman编码,使得出现频率相似的词在隐藏层激活的内容基本一致,而那些出现频率更高的词语激活的隐藏层数目反而更少,这种方法可以有效降低计算复杂度。通过Skip-gram模型分别获取药物分子和蛋白质靶点的低维实数向量特征表示(DrugVec和TargetVec),将原始的非数值数据数值化,以便于用Borderline-SMOTE算法平衡数据集,并且可以直接被分类模型所采用。 采用Word2Vec从文本语料中得到词向量总共需要4步:分词、统计词频、构建Huffman树、输入文本训练词向量。以一个样本(w,context(w))为例,其中context(w)由w前后c个分词组成,输入层只包含了当前样本的中心词w的词向量vw∈。投影层为恒等映射。输出层对应一棵Huffman树,它以语料中出现过的词为叶子节点、以各词在语料库中出现的次数作为权值构造而成。具体于Skip-gram模型,已知当前词w,需要对其上下文context(w)中的词进行预测,所以关键是条件概率p(context(w)|w)的构造,Skip-gram模型将其定义为: (2) Skip-gram模型的算法流程如下所示。 输入:基于Skip-gram的语料训练样本,词向量的维度大小M,上下文窗口大小c,步长η。 输出:Huffman树的内部节点模型参数θ,所有的词向量v(w)。 第一步:基于训练样本建立Huffman树。 第二步:随机初始化Huffman树的内部节点模型参数θ、所有的词向量v(w)。 第三步:进行梯度上升迭代过程,对于训练集中的每一个样本(w,context(w))进行如下处理: 对于每个样本w,从根节点到输入词的节点总数设为lw。在Huffman树中从根节点开始,经过第j (j=2,…,lw)个节点的Huffman编码为dwj; 1)设e=0; 2)对于context(w)中每一个分词u,计算词向量v(w)并更新θ:f=σ(v(w)Tθwj-1)g=η(1-dwj-f)ee+gθwj-1θwj-1θwj-1+gv(w) 3)如果v(w)=v(w)+e梯度收敛,则结束梯度迭代,算法结束,否则回到步骤1)继续迭代。 设置Skip-gram模型的参数时,通过机器学习库scikit-learn中的RandomizedSearchCV方法进行超参数调整,选取最优参数:词嵌入的维度设置为100,上下文窗口大小设置为12,负例数设置为15。进行药物分子和靶点蛋白质特征表示训练,分别获得各100维的药物分子和靶点蛋白质的词向量(DrugVec和TargetVec),将二者组合成200维的DTVec,用以表示药物分子和靶点蛋白质之间的相互作用特征。 选择GBDT作为分类器,对处理之后的数据进行分类预测训练。GBDT中的树都是回归树,主要用于回归预测,可设置阈值将其用于二分类预测(预测结果大于阈值的设为正样本,反之则为负样本)。GBDT中每棵树学习到的都是之前所有树的结果和的残差,通过损失函数的负梯度拟合,使预测值逐步逼近真实值。本文采用的损失函数是对数似然损失函数。 在模型训练过程中进行超参数优化,选取最优参数:弱学习器的最大迭代次数n_estimators设置为60,学习率learning_rate设置为0.2。为了佐证参数的合理性,以Nuclear Receptor数据集为例,绘制损失函数曲线。 n_estimators=60时,学习率learning_rate的损失函数曲线如图1所示。由图1可见,在学习率为0.05左右时,训练集loss值接近最低点并保持相对稳定;在学习率从0增至0.2时,交叉验证集的loss值下降很快,在学习率大于0.2后,loss值下降速率趋缓。为了防止学习率过高导致的过拟合问题,本文设置学习率learning_rate=0.2。 learning_rate=0.2时,弱学习器的最大迭代次数n_estimators的损失函数曲线如图2所示。由图2可见,训练集loss值一直很低,说明模型在训练集上表现良好;在交叉验证集上,loss值开始随n_estimators的增加而减小,在n_estimators约为60时达到最低点,之后loss值随着n_estimators的增加而提高,这是因为迭代次数增多导致模型过拟合,因此设置弱学习器的最大迭代次数n_estimators=60。 图2 n_estimators的损失函数曲线 准确率(ACC)通常是评价机器学习模型的重要指标,但由于本实验数据集的高度不平衡性,ACC不宜作为主要的评价指标。ROC(Receiver Operating Characteristic)曲线以及AUC(Area Under Curve,ROC曲线下的面积)常用来评价针对不平衡数据集的二元分类器的性能。ROC曲线的横轴为假正率(False Positive Rate,FPR),即误判为正样本的比率: (3) ROC曲线的纵轴为真正率(True Positive Rate, TPR),即正确判断为正样本的比率: (4) 式(3)~式(4)中:TP表示真正例;FN表示假负例;FP表示假正例;TN表示真负例。 针对负样本数据缺失的问题,本文首先在未知的药物-靶点相互作用对数据中通过随机采样生成负样本数据。初步设定生成的负样本数量分别为正样本数量的1倍、5倍、10倍,对所生成负样本的实验效果进行了检测,结果发现,生成10倍于正样本的负样本取得的效果最佳,所以最终选取10倍于正样本的未知样本作为负样本。 为了验证本文方法的性能,将其与文献[1]、文献[18-23]中的多种方法进行实验对比,均使用相同的数据集,采用十折交叉验证,在每个数据集上各进行十次实验,根据平均结果计算 AUC值。 图3为本文方法应用于黄金标准数据集得到的ROC曲线和AUC值,可以看出,其在4个数据集上均表现出优异的性能。表2为不同方法在黄金标准数据集上的实验结果比较。Enzyme、GPCR和Ion Channel是数据量比较丰富的3个数据集,对于Enzyme数据集,本文方法略优于AdaBoost和iDTI-ESBoost,明显优于其他对比方法,在GPCR和Ion Channel数据集上,本文方法优势明显。在数据量最小的Nuclear Receptor数据集上,本文方法取得的AUC值为0.9013,逊于AdaBoost和iDTI-ESBoost,但仍优于其他方法。综合来看,本文方法在数据量较多的数据集上表现优异,在数据量少的数据集上也取得不错的结果。因此,本文提出的基于药物结构信息和靶点序列信息的词向量特征提取策略对DTI特征进行表示并针对更具代表性的少数类样本(边界样本)生成新样本的数据预处理方法取得了较好的预测性能,并且能够适用于不同规模的数据集。 (a)Enzyme (b)GPCR (c)Ion Channel (d)Nuclear Receptor 表2 不同方法应用于黄金标准数据集的AUC值对比 表3为不同方法在DrugBank 4.3数据集上的实验结果比较。由表3可见,与同样采用集成学习的RF等方法相比,本文方法在AUC和ACC这两项指标上均较优,并且ACC指标的优势明显。数据集不平衡的特性使得大部分实验模型在ACC值(准确率)上很容易取得好的结果,因此,相关研究不将ACC值作为主要评价指标,但这并不意味着ACC指标不重要。RF、DBN等方法的AUC指标值虽然还比较高,但ACC值均低于0.9,这种以准确率为代价的实验结果证明了RF等方法的不完善。另外,与深度学习方法CNN+sequence进行比较,本文方法在两项指标上均取得更好的实验结果。表3数据进一步说明了本文方法对于不平衡数据处理的有效性。 表3 不同方法应用于DrugBank4.3数据集的AUC、ACC值对比 本文针对药物-靶点相互作用数据集不平衡的问题,提出了一种有效的数据处理方法BS-DTVec。该方法基于Word2Vec模型提取药物和靶点数据的词向量特征,并组合成药物-靶点相互作用特征,同时根据少数类样本中的特定样本(即处在少数类和多数类样本边缘的样本)产生新的少数类样本,缓解了正、负样本数据量的较大差异对分类模型泛化能力的不利影响。词向量特征提取方式可以较好地提取出重要的药物化学结构特征(DrugVec)以及靶点蛋白质氨基酸序列片段特征(TargetVec),其组合(DTVec)可以有效地表示药物-靶点相互作用的关键特征。通过样本空间中的边缘样本生成新的样本,使少数类样本和多数类样本的边界更加明显,避免了因在少数类样本集群内部生成新样本而对模型训练可能造成的过拟合问题。这种数据处理方式更加有助于分类模型在训练过程中区别正样本和负样本。在不同数据集上的实验结果证明了BS-DTVec方法能有效处理药物-靶点相互作用预测数据集不平衡问题。在今后的研究过程中将考虑扩充数据集,针对特定疾病的药物-靶点相互作用进行研究,为具体的疾病治疗提供辅助方案。1.3 特征表示


1.4 分类模型
2 实验
2.1 评价指标
2.2 负样本采样倍率的确定
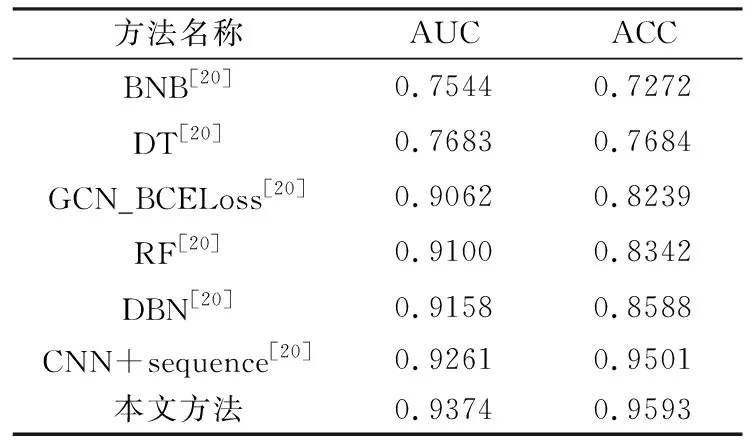
2.3 实验结果评估

3 结语
猜你喜欢
体育科技文献通报(2022年3期)2022-05-23
安徽医科大学学报(2022年4期)2022-05-12
新高考·高一数学(2022年3期)2022-04-28
计算机研究与发展(2022年1期)2022-01-19
中老年保健(2021年3期)2021-12-03
中学生数理化(高中版.高考数学)(2021年1期)2021-03-19
计算机应用(2020年12期)2020-12-31
高中生学习·高三版(2016年9期)2016-05-14
新高考·高二数学(2015年11期)2015-12-23
文苑(2015年9期)2015-09-10
