
低密度脂蛋白受体相关蛋白5调节骨量的作用机制及其影响因素研究进展
2022-11-27 14:42:24牛娜王丽杨娓霞赵乃倩
山东医药 2022年32期
牛娜,王丽,杨娓霞,赵乃倩
1山西医科大学第二临床医学院内分泌科,太原 030001;2山西医科大学第二临床医学院老年科
骨质疏松症是以骨密度降低、骨组织微结构破坏和骨折风险增加为特征的一种多因素疾病。遗传学研究表明,高达80%的骨密度群体变异是由遗传因素决定的[1]。研究显示,低密度脂蛋白受体相关蛋白5(LRP5)基因突变是骨质疏松—假性神经胶质瘤综合征(OPPG)和常染色体显性高骨量表型相关疾病的发病基础[1-2]。LRP5是一种细胞表面信号转导受体,由1 615个氨基酸组成,可分为胞外结构域、跨膜结构域和胞内结构域三部分[1]:氨基端胞外结构域依次由信号肽、四个YWTD型β螺旋桨结构域和三个LDLR配体结合基序组成;跨膜结构域是由23个氨基酸组成的一重跨膜结构域;羧基端胞内结构域含有207个氨基酸,含有16%的脯氨酸残基、15%的丝氨酸残基和5个重复的PPP(S/T)P基序[3-4]。有研究指出,LRP5基因突变可能是骨质疏松症的重要原因[5-6]。在部分原发性骨质疏松症而无眼部病变的儿童和成人患者中,存在LRP5基因杂合错义突变。LRP5单核苷酸多态性与年轻人群的骨密度改变密切相关,并可能最终影响峰值骨量的获得;在老年人群中,LRP5基因突变与骨质疏松性骨折密切相关[7]。因此,LRP5可能是一般人群骨质疏松症的易感基因。了解LRP5调节骨量的机制及其影响因素,有助于开发评估骨质疏松症、骨折风险的遗传标志物,研发防治骨质疏松症和其他低骨量疾病的新方法。现就LRP5调节骨量的作用机制及其影响因素相关研究进展综述如下。
1 LRP5调节骨量的机制
1.1 LRP5激活经典的Wnt/β-catenin信号通路在细胞膜上,配体蛋白质Wnt与LRP5和Frizzled受体家族成员结合,诱导LRP5和Frizzled胞内结构域发生构象变化。变构的LRP5胞内结构域募集Frat1、Axin、结肠腺瘤样息肉(APC)和糖原合成酶激酶(GSK)-3β等胞内蛋白,形成Frat1-Axin-APCGSK-3β蛋白复合体。其中,Axin充当支架结构,Frat1为GSK-3β结合蛋白,可抑制GSK-3β的活性。变构的Frizzled胞内结构域募集Dissheveled,后者也可抑制GSK-3β的活性。GSK-3β活性抑制可抑制β-catenin磷酸化,使得β-catenin不能与Axin、APC和GSK-3β形成Axin-APC-GSK-3β-β-catenin降解蛋白复合体,使β-catenin免于被蛋白酶体降解。β-catenin在细胞质中蓄积并转位至细胞核,与转录因子T细胞因子/淋巴增强因子相互作用,调节碱性磷酸酶和间隙连接蛋白等靶基因的转录[3,8]。LRP5/Wnt/β-catenin信号通路组成及信号转导模式见图1。
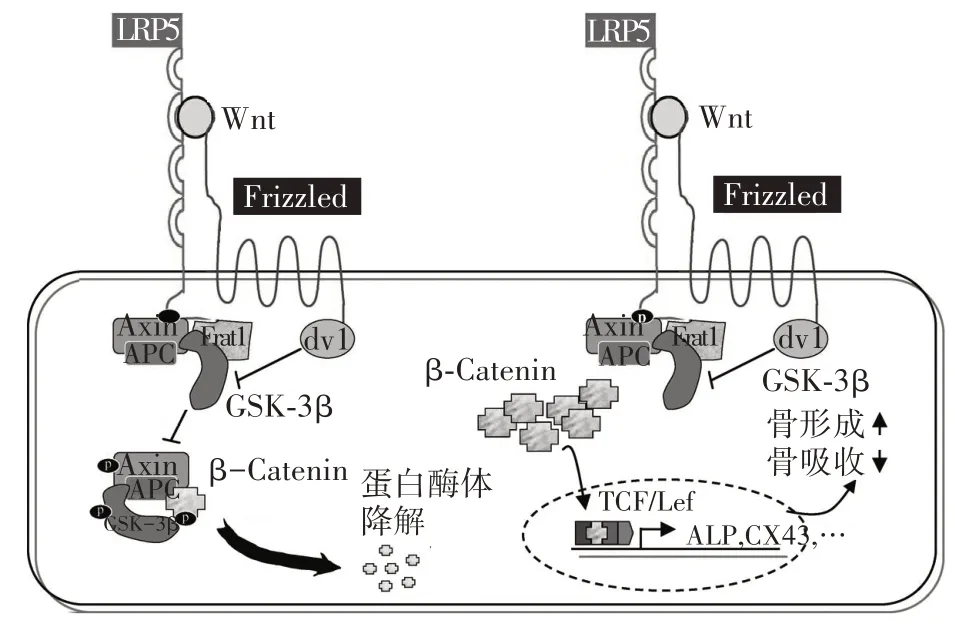
图1 LRP5/Wnt/β-catenin信号通路组成及信号转导模式(改编自参考文献[3])
骨经典Wnt/β-catenin效应的靶细胞主要是成骨细胞,可调节骨内膜和骨髓基质内间充质始祖细胞向成骨细胞定型和分化,并影响成骨细胞的增殖和功能,进而调节骨形成和骨重塑[9]。此外,成骨细胞经典Wnt/β-catenin信号通路的靶基因骨保护素也是NF-κB受体活化因子(RANKL)的诱饵受体,通过与RANKL的相互作用阻断破骨细胞的RANKL骨分解代谢通路,进而抑制破骨细胞的分化和功能。这表明成骨细胞经典Wnt/β-catenin信号通路对破骨细胞的分化和功能具有间接的调节作用[10]。
值得注意的是,成骨细胞经典Wnt/β-catenin效应主导的骨形成和骨重塑过程离不开能量代谢的支持。成骨细胞经典Wnt/β-catenin信号通路的活化可诱导脂肪酸β氧化所需关键酶的表达,通过增强成骨细胞的脂肪酸β氧化促进骨形成和骨重塑,同时伴有全身脂肪酸β氧化增强引起的机体脂肪减少、能量消耗增加及血甘油三酯和游离脂肪酸水平下降等表现[11-12]。LRP5和Wnt也可以β-catenin非依赖的方式激活成骨细胞内重要的信号转导分子Rac1,进而激活其下游的mTORC2/AKT信号转导通路,诱导糖酵解关键酶的表达,通过增强有氧糖酵解促进骨形成和骨重塑,上调血清乳酸水平[11,13]。
1.2 LRP5抑制5-HT/5-HTR1B/CREB信号通路5-羟色胺(5-HT)是一种单胺类神经递质,不能通过血脑屏障,其对骨量的调节作用因合成部位不同而异,中枢部位的5-HT可促进骨形成,而外周部位的5-HT可抑制骨形成[14]。中枢部位的5-HT是由脑干中缝核的突触前神经元合成的,其对骨量的调节作用不依赖于LRP5/5-HT/5-HTR1B/CREB信号通路。大部分外周5-HT是由十二指肠内特化的神经内分泌细胞肠嗜铬细胞合成的[14]。LRP5可抑制肠嗜铬细胞中催化5-HT合成的限速酶色氨酸羟化酶1(TPH1)表达,抑制5-HT的合成,使外周5-HT水平降低。而外周5-HT可与成骨细胞前体细胞上的5-HT受体1B(5-HTR1B)结合,通过调节转录因子叉头框蛋白O1(FOXO1)与cAMP反应元件结合蛋白(CREB)结合及FOXO1与激活转录因子4(ATF4)结合之间的平衡,调节FOXO1的转录活性。FOXO1与CREB结合可促进骨形成,FOXO1与ATF4结合则抑制骨形成。外周5-HT的增加通过抑制FOXO1与CREB的结合,增强FOXO1与ATF4的结合,抑制细胞周期蛋白D1、D2、E3等靶基因表达,从而抑制成骨细胞增殖和骨形成[14-18]。LRP5/5-HT/5-HTR1B/CREB信号通路组成及信号转导模式见图2。
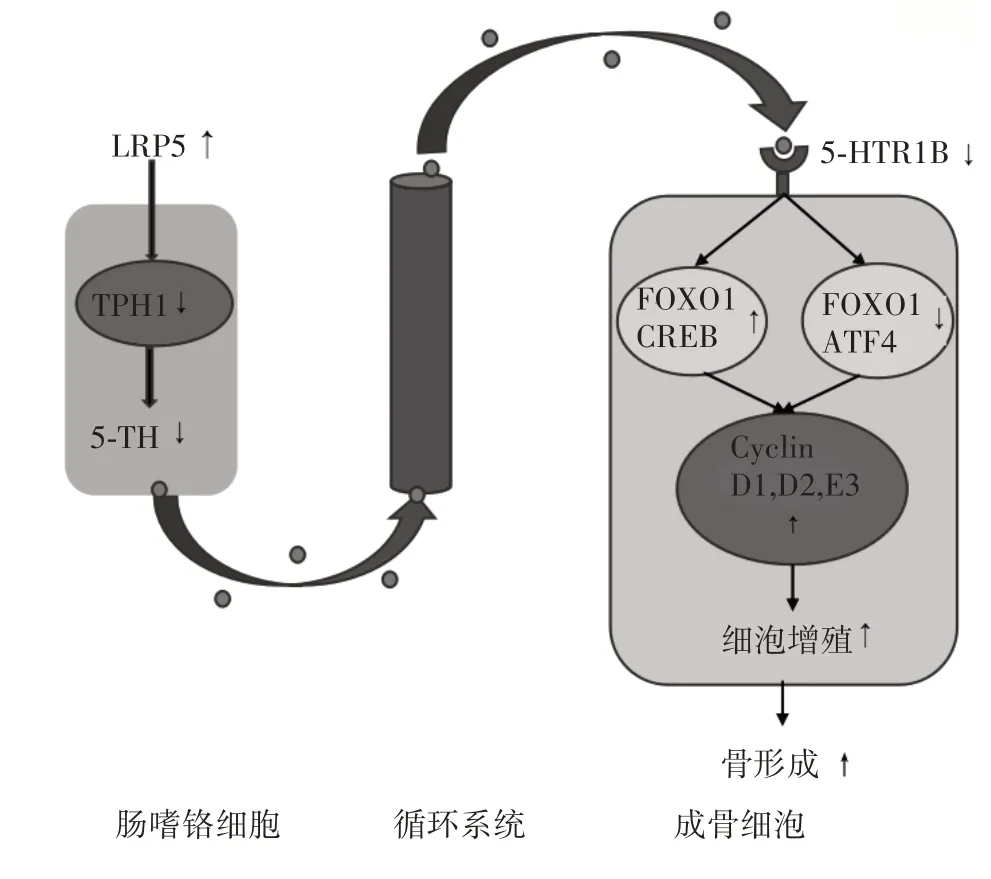
图2 LRP5/5-HT/5-HTR1B/CREB信号通路组成及信号转导模式
2 LRP5调节骨量作用的影响因素
2.1 LRP5/Wnt/β-catenin信号通路的影响因素LRP5是经典Wnt/β-catenin信号通路调节骨量的关键因子,LRP5表达水平会对其骨量调节功能产生影响。LRP5在人结直肠癌组织中的表达受LRP5基因启动子甲基化的调控。肿瘤坏死因子受体相关蛋白1可通过降低LRP5基因启动子甲基化水平从而诱导LRP5表达[19],但这一调控机制在成骨细胞中的作用尚不明确。在LRP5 TSS下游的+19 kb区域有维生素D受体的反应元件(VDRE),VDRE与VDR和维甲酸X受体(RXR)形成的异源二聚体结合后具有基因转录功能。1,25-二羟维生素D3[1,25-(OH)2D3]可通过诱导VDR/RXR异源二聚体的形成,促进成骨细胞LRP5表达[20-21]。
LRP5的不同结构域具有结合不同调节蛋白的特性,这些调节蛋白是调节LRP5/Wnt/β-catenin信号通路的重要因素[1,3,8,22]。Wnt是LRP5的配体,与LRP5的第二β螺旋桨结构域结合以启动信号转导。中胚层发育蛋白(MESD)可能是一种主要与LRP5第一β螺旋桨结构域相互作用的伴侣蛋白,可促进LRP5由细胞质向细胞膜转运,是LRP5定位至细胞膜所必需的。Dickkopf1(DKK1)和骨硬化蛋白(Sclerostin)是两种糖蛋白,DKK1可与LRP5第三β螺旋桨结构域结合,同时与Kremen受体结合形成三聚体,诱导LRP5快速内吞并从质膜上去除,减少细胞膜上的LRP5,抑制Wnt信号向胞内传递。Sclerostin可与LRP5的第一和第二β螺旋桨结构域结合,使Wnt配体与LRP5结合受阻,进而抑制Wnt/βcatenin信号转导。LRP5胞内结构域可结合Axin和Frat1以促进Wnt信号在胞内传递[23]。LRP5胞内结构域中的PPP(S/T)P基序是Axin的结合位点。LRP5羧基末端47个氨基酸构成的区域包含三个重复的PPP(S/T)P基序,但这些PPP(S/T)P基序并不参与LRP5与Frat1的结合,这一区域中1 569~1 588氨基酸残基构成的亚区域是Frat1的结合位点。Frat1可通过使Axin与GSK-3β解离,抑制GSK-3β的活性,进而减少β-catenin的降解。
迄今为止,OPPG相关LRP5功能缺失型突变分布于LRP5蛋白的各个结构域,但超过90%的突变位于胞外结构域[24]。LRP5有害突变可导致体内无法合成LRP5蛋白或合成截短的LRP5蛋白,使Wnt信号无法向胞内传递,进而引起低骨量改变。LRP5有害突变也可损伤LRP5的结构与功能。LRP5结构改变可能影响LRP5与MESD的结合,LRP5不能有效向细胞膜转运,使细胞膜上的LRP5不能达到应有的数量,导致Wnt信号活性减弱。LRP5结构改变还可能影响LRP5与Wnt配体的结合,导致Wnt信号活性减弱。LRP5功能获得型突变均位于第一β螺旋桨结构域,可能通过减少LRP5与Sclerostin或DKK1的结合,削弱Sclerostin或DKK1对Wnt信号的拮抗作用,导致骨形成增加[24]。
2.2 LRP5/5-HT/5-HTR1B/CREB信号通路的影响因素诱导LRP5表达的因素可抑制5-HT/5-HTR1B/CREB信号通路,促进成骨细胞增殖和骨形成。TPH1抑制剂LP533401不易通过血脑屏障,主要作用于肠嗜铬细胞,通过减少外周5-HT的合成促进成骨细胞增殖,从而治疗卵巢切除小鼠和LRP5缺乏小鼠的骨质疏松症[25]。选择性5-HT再摄取抑制剂(SSRIs)通过抑制5-HT转运体、增加5-HT的利用率,导致进入循环的5-HT增加,这可能是使用SSRIs治疗的患者骨折风险增高的原因[26]。除此之外,影响5-HT/5-HTR1B/CREB信号通路的因素还有低色氨酸饮食、5-HT受体拮抗剂和5-HT受体激动剂[16,27],这些因素或通过降低5-HT水平、阻断5-HT的作用,或通过增强5-HT的作用来调节成骨细胞增殖和骨形成,但上述调节作用都不是通过改变LRP5表达或改变LRP5对TPH1表达的调节功能实现的。
研究显示,LRP5纯合功能缺失型突变、复合杂合功能缺失型突变及杂合功能缺失型突变患者在罹患早发性骨质疏松症的同时,血清5-HT水平明显升高[27-30];而LRP5功能获得型突变患者在骨量显著增加的同时,血清5-HT水平明显降低[27,29,31]。这表明LRP5基因突变可能通过改变5-HT/5-HTR1B/CREB信号通路的活性调节骨形成,但LRP5基因突变对5-HT表达的调控机制尚不清楚。
综上所述,LRP5是骨质疏松症的易感基因,能够激活经典Wnt/β-catenin信号通路,抑制5-HT/5-HTR1B/CREB信号通路,从而促进骨形成、增加骨量。人群中存在着复杂的LRP5基因突变[32],这些基因突变可通过多种机制调节经典Wnt/β-catenin信号通路和5-HT/5-HTR1B/CREB信号通路功能[33-34]。然而,每位因LRP5基因突变而发生骨量改变的个体,其背后的发病机制可能都是不一样的,需要在基因型确定后进一步明确其功能和具体的致病机制,开展个体化治疗。随着Sclerostin、DKK1等调节蛋白作用机制的阐明,针对Sclerostin和DKK1的单克隆抗体相继问世,有望为骨质疏松症的治疗提供更多选择[18,35]。此外,LRP5单核苷酸多态性与年轻人群峰值骨量的获得和老年人群骨质疏松性骨折风险密切相关,而1,25-(OH)2D3可诱导LRP5表达,提示对于LRP5功能未完全受损的人群尽早使用维生素D治疗,可能会通过增加LRP5表达进行代偿,从而有效预防和延缓LRP5相关骨质疏松症的发生和发展。
猜你喜欢
云南医药(2021年3期)2021-07-21 05:40:20
实用医药杂志(2020年8期)2020-08-26 01:59:36
现代临床医学(2019年6期)2019-12-07 06:03:12
安徽医科大学学报(2016年12期)2017-01-15 14:21:53
中国民族医药杂志(2016年6期)2016-05-09 08:52:52
中国医科大学学报(2015年10期)2015-03-01 02:09:58
中国医药导报(2015年24期)2015-02-28 22:07:30
现代检验医学杂志(2014年2期)2014-02-02 02:40:54
广州体育学院学报(2014年6期)2014-01-31 02:36:36
中国医学科学院学报(2011年5期)2011-12-01 03:52:51
