
LiCl对聚丁内酰胺的热塑改性研究
2022-11-26 11:22蔡智立张雅敬辛瑞杰邱勇隽蒋丽华赵黎明
功能高分子学报 2022年6期
蔡智立, 张雅敬, 辛瑞杰, 邱勇隽, 陈 涛, 蒋丽华, 赵黎明
(华东理工大学 1. 生物工程学院,生物反应器工程国家重点实验室;2. 中国轻工业生物基材料工程重点实验室;3. 材料科学与工程学院,上海市先进聚合物材料重点实验室 , 上海 200237)
聚丁内酰胺(Polybutyrolactam),又叫聚酰胺4(Polyamide 4,PA4),是以丁内酰胺为原料,经阴离子开环聚合得到的一种乳白色聚酰胺,丁内酰胺可由生物基来源的γ-氨基丁酸脱水环化制备。作为聚酰胺家族中的一员,PA4具有优良的力学性能,在耐酸、耐碱、耐磨和抗静电等方面也比其他热塑性树脂表现优异。另外,PA4在土壤生物堆肥[1]、活性淤泥[2]、海水[3]和生物体内[4]表现出优异的生物降解性,是目前唯一一种生物基可降解聚酰胺材料。由于PA4分子链中只含有3个亚甲基重复单元,所以其氢键密度比尼龙6和尼龙66等长链聚酰胺的大,导致其熔点高,且熔点与其分解温度接近,使得热加工比较困难,限制了PA4的商业化应用。目前,PA4的加工手段主要是静电纺丝。Lenka等[5]以甲酸为溶剂,通过静电纺丝成功制备了直径为100 nm的优质纳米纤维,并制备了连续纤维层。还有研究人员将PA4和聚乳酸(PLLA)的嵌段共聚物以13%的质量分数溶解在六氟异丙醇溶剂中,制备了PLLA-b-PA4静电纺丝纤维[6]。曾贝迪等[7]以PA4和壳聚糖(CS)为原料,以甲酸为溶剂制备了PA4/CS电纺纳米蛛网复合纤维膜。虽然静电纺丝可避免PA4的热分解,但是该加工方法对设备要求较高,同时需要大量挥发性有毒溶剂,存在生产效率低、操作环境差和加工成本高等缺点。
为了提高PA4的热加工性能,国内外研究人员开展了一些探索。从结构角度,Koichiro等[8]用羧基、氨基和烷基修饰PA4的末端,结果显示,末端被上述3种基团取代的PA4其热分解温度有所提高,但该方法操作十分复杂,不具有推广性。此外还有研究表明,具有长链脂肪酸基团末端的PA4能够抑制其固有的降解性能[9]。Kawasaki等[10]用羟甲基取代酰胺键上的氢来制备熔点较低的PA4,以改善PA4的热稳定性,但随着羟甲基取代度的增加,改性PA4的生物降解率下降,当取代度为36.5%时,改性PA4在30 d内不发生任何降解。虽然通过结构改性能够在一定程度上提高PA4的热稳定性,但至今鲜有实现PA4熔融加工的有效方法。
本文采用熔融共混挤出的方式,在PA4熔融加工过程中添加LiCl、增塑剂月桂内酰胺和抗氧剂1 010,在双螺杆挤出机中进行熔融挤出制得改性PA4(LiCl/PA4)。相比于PA4分子结构上的改性方法,采用此方法不仅操作简单,还能在不改变其主链结构的情况下有效降低PA4的热加工成本并拓宽其应用领域。该方法在保证PA4降解性能的基础上为其实现商业化热加工及应用提供了有效路径。
1 实验部分
1.1 原料和试剂
聚丁内酰胺(PA4):Mη=2×104,实验室自制,将自制的PA4放入80 ℃的鼓风干燥箱中烘24 h,再将PA4粉末放入105 ℃的真空干燥箱中干燥24 h后备用;氯化锂(LiCl):w≥99%,上海阿拉丁生化科技股份有限公司;甲酸(FA):w≥88%,上海泰坦科技有限公司;月桂内酰胺:w≥99%,上海耐澄生物科技有限公司,放入80 ℃的真空干燥箱中干燥6 h后备用;抗氧剂1010:w≥99%,巴斯夫(中国)有限公司。
1.2 实验步骤
母粒的制备:称取10 g的LiCl溶于适量水中,另称取20 g的PA4溶解于适量的甲酸中,待两者充分溶解后混合在一起,充分反应24 h后,将混合溶液放在40 ℃的加热板上加热,待溶剂挥发完后,将干物质放在105 ℃的真空干燥箱进行烘干,恒重后产物即为母粒。
熔融挤出:将干燥好的PA4粉末、母粒、月桂内酰胺(w=8%)、抗氧剂1010(w=0.6%)按照一定的比例放入高速搅拌机中混合均匀,然后将混合物放入双螺杆挤出机中进行挤出加工,最后注塑成标准拉伸样条。当LiCl与PA4的质量比分别为0、1%、2%、3%、4%、5%时,对应的样品分别标记为S-0(空白对照样品)、LiCl/PA4-1、LiCl/PA4-2、LiCl/PA4-3、LiCl/PA4-4、LiCl/PA4-5。
1.3 测试与表征
广角X射线衍射(XRD)仪:日本理学公司D/max 2550 VB/PC型,放射源为 Cu 靶 Kα射线,40 kV 工作电压,40 mA 工作电流,扫描速率为 5(°)/min,扫描范围为5°~75°。
差示扫描量热(DSC)仪:美国TA仪器公司modulated DSC2910 1090 B型,取适量(6~8 mg)的样品,在N2氛围下,以10 ℃/min的升温速率从25 ℃升至278 ℃。
X射线光电子能谱(XPS)仪:英国Thermo Fisher公司ESCALAB 250 Xi型,使用单色化的 Al Kα 射线源(1 486.6 eV),参照样品表面上自带的空气污染碳化合物的 C1s 峰(284.8 eV)对每个元素窄扫谱图进行能量位移校正。样品的长为5 mm,宽为3 mm,厚为2 mm。

表 1 LiCl/PA4的各项XRD参数Table 1 Various XRD parameters of LiCl/PA4

图 1 LiCl/PA4的XRD谱图Fig. 1 XRD patterns of LiCl/PA4
傅里叶红外光谱(FT-IR)仪:美国热电公司Nicolet is50型,将样品压薄,采用衰减全反射,扫描范围为4 000~400 cm-1,扫描次数为16次。
万能拉力试验机:深圳新三思材料检测有限公司2 T/CMT 4204型,按GB/T 1040—2018进行测试,拉伸速率为20 mm/min。
2 结果与讨论
2.1 LiCl/PA4的结晶度
图1所示是LiCl/PA4的XRD谱图。由图1可知,LiCl/PA4在衍射角(2θ)20.7°和24.0°附近出现2个α晶型的衍射峰,分别对应200晶面和020晶面,在2θ为18.5°处的衍射峰代表PA4的012晶面[11]。图1中并未出现LiCl的衍射峰[12],说明改性PA4体系中LiCl不是以独立的晶体形式插入。随着LiCl含量的增加,020晶面的峰强度显著减弱。200晶面的位置发生了一定程度的偏移,即晶面间距先减小后增大,但是改性后的PA4没有新晶型产生。200晶面间距和020晶面间距分别代表PA4分子中氢键片内的链间距离和片间距离,其中200晶面是通过氢键连接,其晶面间距和PA4氢键的强弱有关[11]。如表1所示,LiCl/PA4-5的200晶面间距增大, 而020晶面间距也有所变化,结晶度下降,这是因为LiCl可以插入PA4的分子链中,使晶面间距增大,破坏了PA4原分子间的氢键,整体上抑制了PA4的结晶[13]。由图1可知,在加入LiCl 后,012 晶面对应的衍射峰消失,这是因为LiCl 破坏了PA4 分子链的原有构象,使012 晶面不能稳定存在,因此 XRD 检测不到对应的衍射峰。采用Jade软件对衍射谱图进行拟合得出的结晶度(Xc)结果列在表1中,随着LiCl含量的增加,改性PA4的结晶度也随之降低,其中LiCl/PA4-5的结晶度最小(37.88%)。这是因为随着Li+含量的增加,与酰胺键(路易斯碱)络合配位增加,对氢键的破坏程度也逐渐增强[14]。
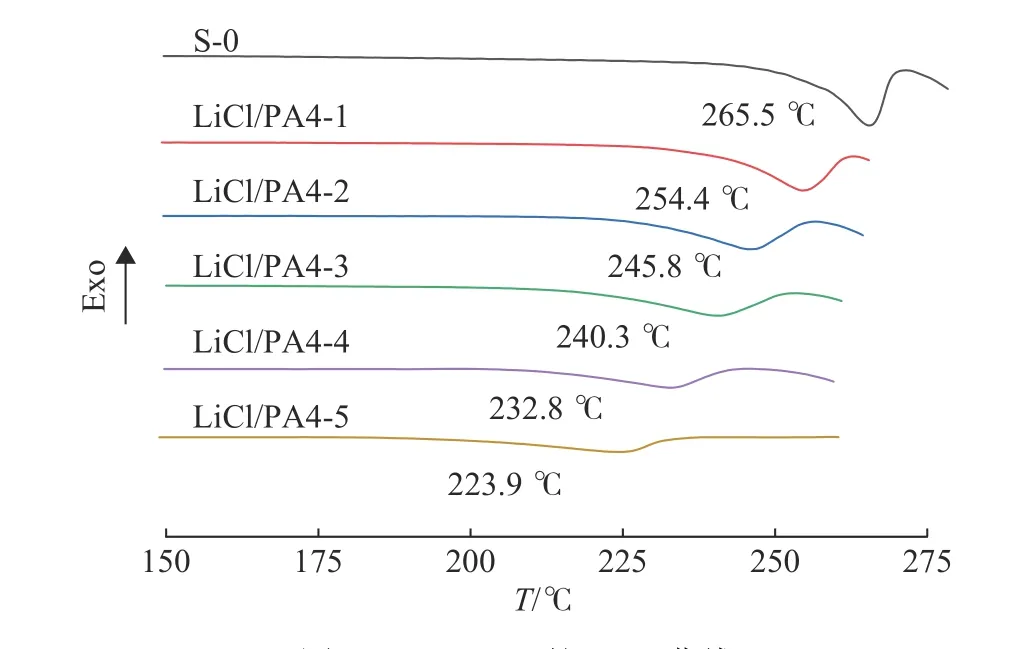
图 2 LiCl/PA4的DSC曲线Fig. 2 DSC curves of LiCl/PA4
2.2 LiCl/PA4的熔融温度
LiCl/PA4的DSC曲线如图2所示。未加入LiCl时,PA4的熔融温度为265.5 ℃。随着 LiCl用量的增加,LiCl/PA4体系的熔点逐渐降低,其中LiCl/PA4-5的熔点降低幅度最大,相比于空白对照样品,熔点降低了41.6 ℃。同时,LiCl/PA4体系的峰面积逐渐减小,无定型区域增多,所以晶体缺陷也越多[15],这也和XRD得出的结论一致。这可能是由于LiCl和PA4发生了络合作用,导致PA4中的氢键被破坏[16]。
2.3 LiCl/PA4的X射线能谱
图3是LiCl/PA4的Li1s、O1s、N1s轨道的X射线能谱图。LiCl/PA4-5的Li1s结合能谱(图3(a))中能够检测出Li元素,表明在熔融挤出过程中,LiCl成功加入到PA4中。LiCl/PA4中Li1s的结合能为55.17 eV,有研究表明无机盐LiCl中的Li1s的结合能为56.10 eV[17]。所以,相比于LiCl样品,LiCl/PA4体系中的 Li1s结合能降低。另外,如图3(b)所示,未增塑PA4的O原子结合能为531.40 eV,将LiCl加入到PA4中,O原子的结合能升高了0.25 eV。这是因为O原子为Li+的空轨道提供孤对电子,产生配位作用,电子转移使得Li原子周围电子云密度升高,屏蔽作用增大,从而使得Li原子内层电子的结合能降低。同时,电子转移使得O原子周围的电子云密度降低,屏蔽作用减小,O原子内层电子的结合能增加[18]。由图3(c)的N1s结合能谱图可知,将LiCl加入到PA4中,N原子的结合能几乎没有变化。因此,LiCl 在PA4中主要是和酰胺基团中的O原子发生络合作用,而不是和N原子发生配位作用。

图 3 LiCl/PA4的XPS 谱图Fig. 3 XPS spectra of LiCl/PA4
2.4 LiCl/PA4的红外光谱
LiCl/PA4的红外光谱如图4所示。波数为3 293 cm-1和3 066 cm-1处分别代表PA4本体的N-H缔合和酰胺Ⅱ带中胺基伸缩振动吸收峰,酰胺Ⅰ带和酰胺Ⅱ带的出峰位置则分别在1 632 cm-1和1 537 cm-1处,相比于未添加LiCl的PA4,添加LiCl的PA4其N-H缔合峰轻微红移至3 291 cm-1,酰胺Ⅱ带中胺基伸缩振动的吸收峰蓝移至3 069 cm-1,而酰胺Ⅰ带和酰胺Ⅱ带分别发生红移(1 630 cm-1)和蓝移(1 539 cm-1),这和前人研究的络合结果一致[19]。
这是由于Li+与C=O形成络合物,破坏了原PA4中的C=O和N-H形成的氢键,束缚了C=O的运动,造成C=O伸缩振动吸收峰(酰胺I带)移至更低的频率,所以发生了红移[20]。而N-H键是自由的,导致N-H伸缩振动带从低频率移向较高的频率发生蓝移;此时,由于氢键被破坏一部分,导致N-H缔合振动频率减弱,但是Cl-会和游离出来的N-H重新缔合,故发生轻微的红移。Li+与C=O的络合作用使得N原子上的电子向C=O转移,增强了C-N键,从而使得酰胺Ⅱ带发生蓝移。
2.5 LiCl/PA4的力学性能
图5所示是LiCl/PA4的断裂伸长率和拉伸强度的变化规律。由图5可知,随着LiCl含量的增加,LiCl/PA4的断裂伸长率增大,其中LiCl/PA4-5的断裂伸长率达到最大值233%,较未加入LiCl的PA4增加了155%,此时柔韧性最好。这是由于LiCl的加入使得Li+与C=O形成络合物,氢键的断裂使分子间的相互作用遭到破坏,结晶度降低,因此PA4中分子链的有序性下降,断裂伸长率增加[21]。
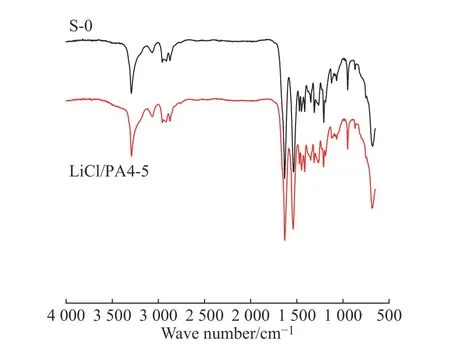
图 4 LiCl/PA4 的FT-IR谱图Fig. 4 FT-IR spectra of LiCl/PA4

图 5 LiCl/PA4的力学性能Fig. 5 Mechanical properties of LiCl/PA4
此外,随着LiCl用量的增加,LiCl/PA4的拉伸强度呈现先增大后减小的变化趋势,平均拉伸强度由S-0的48.76 MPa升高到LiCl/PA-1的57.90 MPa,当LiCl用量继续增加时,拉伸强度减小,LiCl/PA4-5的拉伸强度降至最低为49.14 MPa,但仍然高于空白对照样品。这是由于PA4的C=O与Li+的络合作用是一种强于纯PA4分子间氢键的作用力,LiCl的加入使得PA4分子之间产生交联[22],当LiCl用量较小时,表现出拉伸强度增加的现象,当LiCl用量继续增大时,LiCl/PA4的结晶度降低,氢键破坏程度较高,使得拉伸强度降低。
3 结 论
(1)LiCl的加入使PA4的熔融温度降低,其中LiCl/PA4-5的熔融温度为223.9 ℃。
(2)Li+主要是通过和C=O中的O原子配位破坏PA4分子间的氢键,从而降低LiCl/PA4的结晶度。
(3)LiCl的加入使得LiCl/PA4的断裂伸长率增加,拉伸强度呈现先增大后减小的趋势,但是仍然高于不添加LiCl的PA4,PA4的力学性能显著提高。
猜你喜欢
火炸药学报(2022年5期)2022-11-04
大学物理(2022年9期)2022-09-28
中国现代中药(2022年4期)2022-05-08
中国食品(2020年18期)2020-10-15
物理通报(2020年7期)2020-07-01
物理实验(2019年7期)2019-08-06
航空材料学报(2019年2期)2019-04-15
物理学报(2018年22期)2018-12-18
中国兽医杂志(2016年5期)2016-06-27
原子与分子物理学报(2015年3期)2015-11-24
