
一例BBS12基因复合杂合突变导致Bardet-Biedl综合征的诊断和基因检测分析
2022-11-21 03:22沈艳婷凌雁陆志强李晓牧卞华颜红梅夏明锋常新霞蒋晶晶张晶高鑫
遗传 2022年10期
沈艳婷,凌雁,陆志强,李晓牧,卞华,颜红梅,夏明锋,常新霞,蒋晶晶,张晶,高鑫
遗传资源
一例基因复合杂合突变导致Bardet-Biedl综合征的诊断和基因检测分析
沈艳婷1,2,凌雁1,2,陆志强1,2,李晓牧1,2,卞华1,2,颜红梅1,2,夏明锋1,2,常新霞1,2,蒋晶晶1,2,张晶1,2,高鑫1,2
1. 复旦大学附属中山医院内分泌科,上海 200032 2. 复旦大学慢性代谢性疾病研究所,上海 200032
Bardet-Biedl综合征(Bardet-Biedl syndrome, BBS)是一种罕见的常染色体隐性遗传的纤毛相关疾病,其病因主要与编码BBS蛋白复合体BBSome、鞭毛内运输复合体等基因突变有关。本文报道了1例21岁的女性BBS患者,该患者具有肥胖、视网膜色素变性、双肾囊肿的典型特征,还存在2型糖尿病、非酒精性脂肪肝、亚临床甲状腺功能减退症、轻度传导性听力下降等不典型表现。全外显子组测序发现该患者的基因2号外显子存在复合杂合突变(c.188delC, p.T63fs和c.1993_1995del, p.665_665del)。进一步通过Sanger测序发现患者的父亲和母亲分别携带c.188delC(p.T63fs)和c.1993_1995del(p.665_665del)突变,但均无相关症状。综上所述,本病例报告发现了基因的两个新的突变位点(c.188delC, p.T63fs和c.1993_1995del, p.665_665del),为该疾病的研究提供了新的遗传资源,同时该病例还展示了患者从出生到成人期间的整个疾病发展过程,能够帮助临床医生更好地理解BBS。
Bardet-Biedl综合征;基因;全外显子组测序
Bardet-Biedl综合征(Bardet-Biedl syndrome, BBS)是一种罕见的常染色体隐性遗传的纤毛功能障碍疾病。BBS临床表现多种多样,主要临床特征有肥胖、视网膜色素变性、多指/趾、智力发育迟缓、性腺发育不良、肾脏结构和/或功能异常,次要临床特征主要有肝功能异常、心血管异常、糖代谢异常、神经系统异常、甲状腺功能异常、听力嗅觉异常等。BBS的临床诊断需要符合至少4个主要临床特征或者3个主要临床特征加2个次要临床特征[1]。已有的报道表明,BBS在不同人群中的患病率差异较大,欧洲人群中BBS的患病率在1∶160,000~1∶125,000之间,阿拉伯人群中BBS的患病率为1∶65,000,而贝都因人中BBS的患病率为1∶13,500[2~4]。目前研究发现至少26个基因与BBS的发生发展相关,这些基因通常与纤毛的结构或功能相关[5,6]。其中,基因突变导致的BBS病例约占所有BBS病例的8%~11%,但在中国仅报道了少数病例,我国报道的两例BBS患儿中基因致病突变位点包括c.1604T>G、c.173delA、c.1783T>C和c.1749_ 1750delA等[7~9]。本文报告了1例具有基因两个新突变(c.188delC, p.T63fs和c.1993_1995del, p.665_665del)的女性BBS患者,展示了该患者从出生到成人期间的整个疾病发展过程,不仅为该疾病的研究提供了新的遗传资源,而且有助于BBS的早期诊断和加深临床医生对BBS的理解。
1 对象与方法
1.1 对象及临床资料收集
患者为女性,21岁,汉族,因肥胖自2011年起反复就诊于复旦大学附属中山医院内分泌科,收集患者的病史、体格检查、实验室检查、影像学检查等临床资料,并对患者及其父母进行基因检测。本研究获得复旦大学附属中山医院医学伦理委员会批准,患者及其父母均签署知情同意书。
1.2 外周血DNA提取
采集患者和患者父母EDTA抗凝血3 mL,采用RelaxGene Blood DNA System (北京天根生化科技有限公司)试剂盒提取外周血白细胞基因组DNA。
1.3 全外显子组测序
患者的基因组DNA首先进行全外显子测序。使用Quant-iTTMPicoGreenTMdsDNA试剂盒(Thermo Fisher, 美国)对基因组DNA进行定量。使用NimbleGen SeqCap EZ Human exome Library v3.0 enrichment试剂盒在Illumina HiSeq x-10测序仪上对基因组DNA进行全外显子测序。测序完成后,使用bcl2fastq软件(Illumina)将测序得到的原始数据basecall文件分割转换成fastq文件,将得到的fastq文件通过the Burrows-Wheeler Aligner (BWA)比对软件与人的参考基因组(HG19)进行比对。使用GATK工具InDelRealigner和BaseRecalibrator进行局部重比对和碱基重校正,使用GATK-Unified Genotyper进行基因组变异检测。
1.4 Sanger测序与蛋白质二级结构预测
全外显子组测序检测到的变异通过Sanger测序在患者及其父母的外周血DNA样本进行验证。设计针对检测基因变异的引物:5ʹ-CATGGTGATGGTTGCAGGT-3ʹ(正向)和5ʹ-CATCTTCACTGCACTGCTCC-3ʹ(反向);5ʹ-TTGGCTGGCTTCTCTGG-3ʹ(正向)和5ʹ-AGCTGGGCATTTTAGGCACCA-3ʹ(反向),进行聚合酶链反应(PCR)。PCR扩增条件:95℃ 3 min;95℃ 30 s,30个循环;58℃30 s;72℃ 30 s;72℃ 3 min。采用BigDye Terminator v3.1试剂盒对PCR产物进行测序,利用ABI Prism 3730xl Genetic Analyzer软件对Sanger测序数据进行分析。在OMIM、ClinVar、人类基因突变数据库(Human Gene Mutation Database, HGMD)、Orphanet、GARD等数据库中检索变异是否为已知的致病变异以及是否收录。使用Mutation Taster软件及Prosite预测突变位点的功能。使用SWISSMODEL蛋白质结构建模工具(www.swissmodel. expasy.org)预测蛋白的二级结构。根据美国医学遗传学会(American College of Medical Genetics, ACMG)遗传变异分类标准将检测到的变异分类为“致病的”、“可能致病的”和“意义不明确的”。
2 结果与分析
2.1 BBS患者病史及临床表现
患者出生时体重为3 kg,此后患者的体重不断增加,12月龄时体重达15 kg,21岁时体重为126.5 kg,BMI47.6 kg/m2(尽管期间患者进行了饮食控制、运动和胰高血糖素样肽-1受体激动剂等治疗)。患者7岁时首次发现高甘油三酯血症,8岁时实验室检查发现肝功能异常(丙氨酸氨基转移酶和门冬氨酸氨基转移酶均升高),同时肝脏超声发现肝脏脂肪浸润,随后患者肝脏脂肪含量逐渐增加,诊断为非酒精性脂肪肝。14岁时诊断为2型糖尿病,随后通过饮食控制和二甲双胍治疗,血糖控制良好,糖化血红蛋白稳定在6%左右。患者15岁时质子磁共振波谱(1H-magnetic resonance spectroscopy,1H-MRS)检测肝脏脂肪含量为40.4%。16岁时患者的肝脏脂肪含量达到最高值,为52.96%,此后予以低热量饮食、二甲双胍、小檗碱规律治疗,患者肝功能逐渐恢复正常,18岁时肝脏脂肪含量降至32.2%。
患者在3岁时开始出现视物模糊,症状进行性加重,最终在11岁时诊断为双眼无色素性视网膜色素变性,此时患者双侧裸眼视力已降至<0.1。患者15岁时超声检查显示双侧肾囊肿,肾功能在正常范围,同时诊断为亚临床甲状腺功能减退症,未行甲状腺激素替代治疗,后来规律随访患者的促甲状腺激素水平一直在5 μIU/mL左右(参考范围0.27~ 4.2 μIU/mL)。17岁时患者出现了轻度传导性听力下降。患者没有多指/趾、性腺发育异常、认知或智力障碍、神经系统异常、心血管系统异常等其他临床表现(图1)。
2.2 基因检测结果分析
全外显子组测序发现了该患者具有2个既往未报道的基因的新突变(c.188delC, p.T63fs和c.1993_1995del, p.665_665del),两个突变均位于基因的2号外显子,Sanger测序进一步确认了上述突变(图2A)。该患者的父亲和母亲分别携带了基因的c.188delC(p.T63fs)突变和c.1993_ 1995del(p.665_665del)突变(图2A),但该患者的父母没有BBS相关的症状。c.188delC (p.T63fs)突变是移码突变,在编码区188号上的C碱基缺失,使第63号氨基酸上提前出现终止密码子从而导致转录/翻译提前终止,Mutation Taster软件预测该突变可能影响蛋白功能。基因编码的蛋白包含顶端结构域、中间结构域和赤道结构域,这3个结构域在BBS蛋白中具有保守性,通过Prosite分析显示c.188delC (p.T63fs)能够导致中间结构域、赤道结构域和部分顶端结构域缺失,预测该变异会影响蛋白功能。根据ACMG相关指南,该突变归类为可能致病的变异。而c.1993_1995del(p.665_665del)突变表示在编码区第1993~1995号上存在GTT缺失,从而导致赤道结构域上的第655号缬氨酸缺失,通过Prosite分析显第665位缬基酸不在酪蛋白激酶II磷酸化位点、蛋白激酶C磷酸化位点、N-肉豆蔻酰化位点或N-糖基化位点。Mutation Taster软件预测该突变可能不损害蛋白功能。根据ACMG相关指南,该突变归类为意义不明确的变异。上述两个基因变异位点在OMIM、ClinVar、人类基因突变数据库(HGMD)、Orphanet、GARD数据库中均未被收录。经过生物信息学预测,这两种突变均能改变BBS12蛋白的二级结构(图2B)。此外,该患者未发现其他BBS相关基因或其他纤毛相关疾病基因的变异。
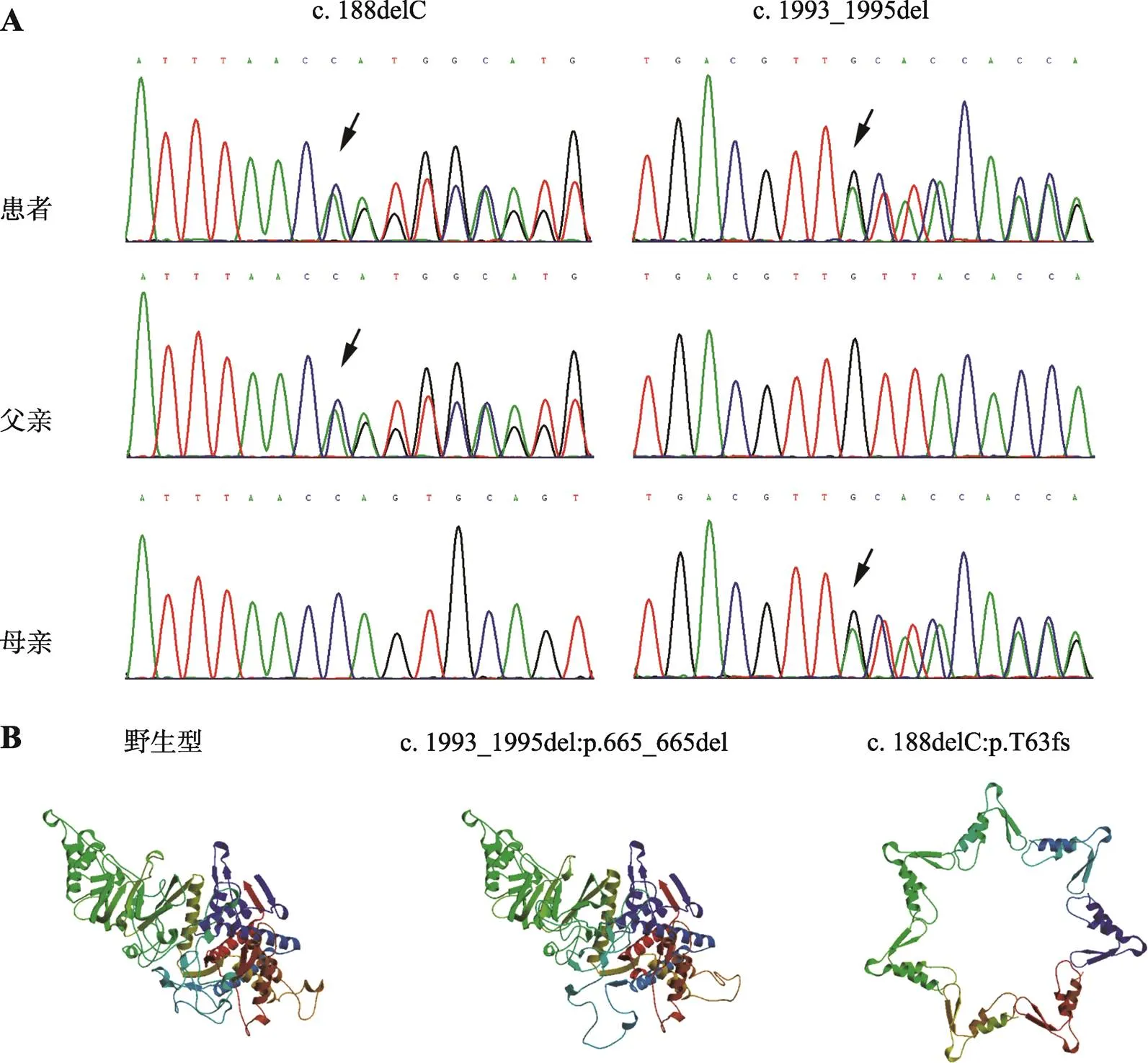
图2 患者及患者父母的基因检测结果及分析
A:在患者和患者父母的外周血DNA中通过Sanger测序验证基因复合杂合突变(c.188delC, p.T63fs和c.1993_1995del, p.665_ 665del)。患者和患者父亲具有c.188delC突变,而患者和患者母亲具有c.1993_1995del突变。B:BBS蛋白的二级结构预测。
3 讨论
BBS是由于基因突变引起的原发性纤毛功能障碍性疾病[10]。相关基因编码的BBS蛋白(BBS1、BBS2、BBS4、BBS5、BBS7、BBS8、BBS9和BBS18)可以形成BBSome复合体,参与纤毛的形成或者一些特定蛋白如G蛋白偶联受体、生长抑素受体的跨纤毛运输,而BBS6、BBS10和BBS12组成的复合物则是BBSome复合体形成过程中必须的伴侣蛋白复合物[11~13]。BBS相关基因突变后可以引起非运动性纤毛功能障碍,常常累及多个器官系统,可以表现出多种临床症状,不同的患者之间临床表现可能存在很大的差异。在BBS的病程中,并不是所有症状都在同一时间出现,相应症状可以在患者婴幼儿时期或者10岁后逐渐出现。此外,纤毛相关疾病还存在着一些重叠的临床表现和致病基因,如Alstrom综合征、Jeune综合征、Joubert综合征和McKusick- Kaufman综合征等[14,15]。例如,肥胖和视网膜色素变性是BBS和Alstrom综合征共有的临床表现,而一些导致McKusick-Kaufman综合征的基因突变也可以导致BBS。因此现有的BBS临床诊断标准并不适用于所有的BBS患者,特别是具有不典型症状的患者[16]。有时BBS的早期确诊存在着困难,需要基因检测才能做出正确的诊断[17]。本例患者基于其目前的临床表现尚不能确诊为BBS,并且该患者还需要进一步与Alstrom综合征等疾病进行鉴别,因此本研究对该患者进行了全外显子组测序,结果发现该患者携带了两个基因的新突变(c.188delC, p.T63fs和c.1993_1995del, p.665_665del),从而确诊为BBS。
基因位于人染色体4q27上,是由两个外显子编码710个氨基酸的蛋白,基因的功能缺失型突变可以导致BBS[18]。迄今为止,尽管在BBS患者中已经报道了60多个基因的突变位点[7, 19],基因型和BBS表型的关系尚待进一步研究。有研究发现,与基因突变的BBS患者相比,基因突变的BBS患者出现认知障碍的频率更高,并且更容易表现出其他严重表型,比如肥胖、多指/趾、短指、心血管系统异常、精神运动迟缓和行为问题等[20]。然而,也有研究报道了相反的结论,基因突变的相关表型更加轻微[16,21]。表1比较了本例患者的表型和已报道的突变患者的表型。
本例患者最主要的症状是严重且早发的视网膜色素变性和肥胖,而上述症状在所有BBS患者中的发生率分别为90%以上和72%~92%[17]。BBS患者早期可出现视杆细胞/视椎细胞的凋亡,早期表现为黄斑受累的非典型色素性视网膜变性,逐渐出现夜盲症,随后出现畏光以及色觉丧失[22,23],症状通常在10岁时出现,大多数患者在20岁或30岁时发展为失明[24]。本例患者在3岁时出现视物模糊,11岁时诊断为视网膜色素变性并且视力已下降至光感,眼部病情是不断进展的。有文献报道大部分BBS患者在出生时体重正常,然而在儿童早期体重可迅速增长,且患有BBS的男性在青少年期BMI可以有改善,但女性则保持不变[25]。肥胖会增加BBS患者的健康问题,包括心血管疾病、阻塞性睡眠呼吸暂停、非酒精性脂肪肝、糖尿病等。本例患者出生时体重正常,12月龄时体重已快速增长至15 kg,在各种体重管理措施下仍然控制不佳,BMI高达47.6 kg/m2,提示BBS的体重管理仍是临床上需要解决的问题。此外,BBS中肾脏疾病的患病率为53%~82%,且突变的患者可能存在更加严重的肾脏疾病[26~28]。一项包含350名患者的研究显示,50%的患者可以发展为功能性肾脏疾病,约8%的患者会继续发展为需要透析或移植的终末期肾脏疾病[26]。不过本例患者虽然有双侧肾囊肿,但到目前为止肾功能仍然在正常范围内。
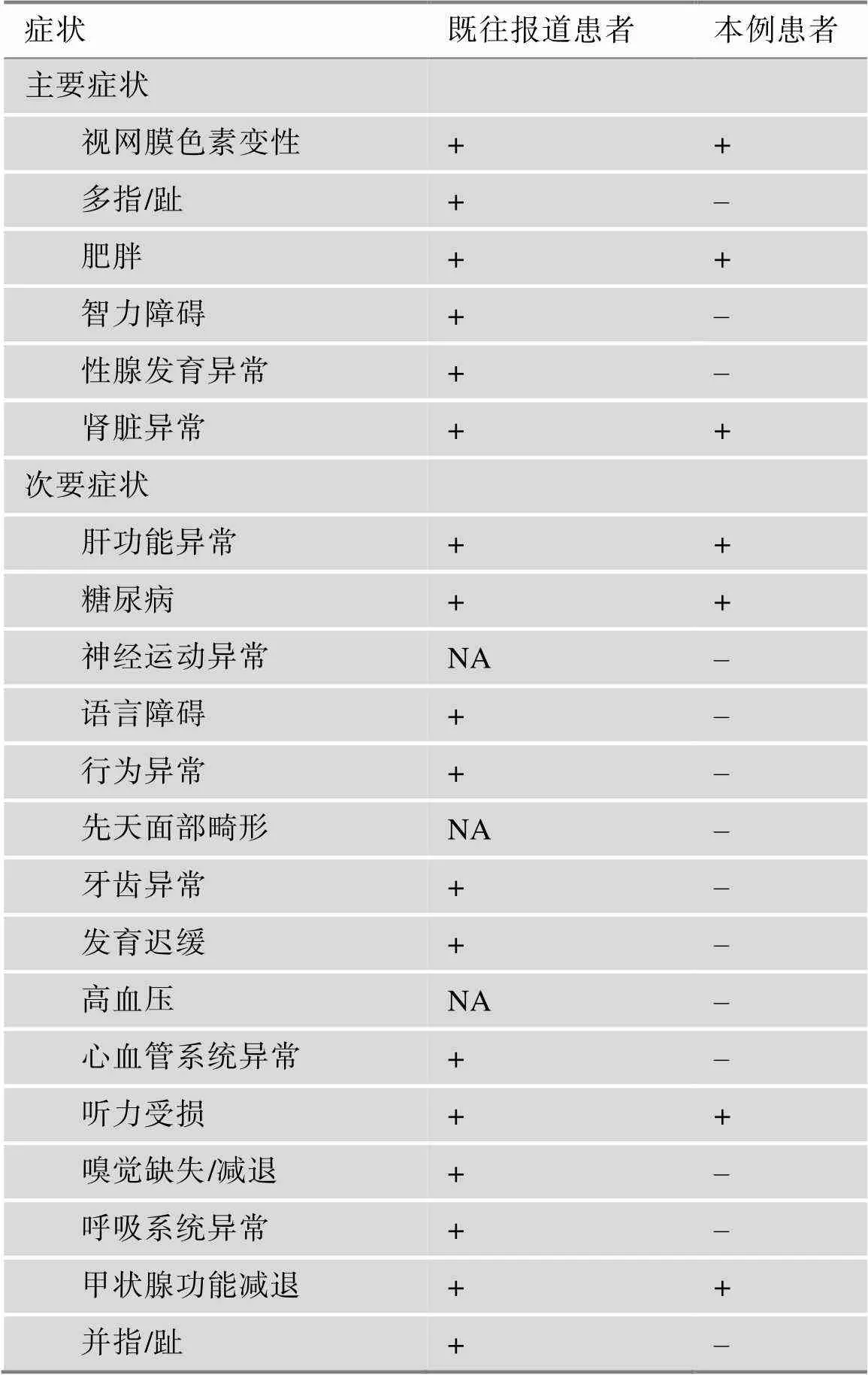
表1 已报道的BBS12基因突变患者的表型与本例患者表型的比较
+:存在;–:未报道;NA:未明确。
本例患者在14岁时被诊断为2型糖尿病,这与BBS患者的2型糖尿病多在青春期发病的报道相一致[17]。有研究表明BBS患者的胰岛素抵抗更普遍[8]。BBS患者的胰岛素抵抗可能与BBS蛋白在胰岛素受体运输和定位中的重要性有关。既往研究表明初级纤毛与胰岛素分泌、胰岛素信号传导和葡萄糖代谢密切相关,而β细胞纤毛功能障碍与2型糖尿病的发生有关[29,30]。然而,也有研究发现的失活突变与胰岛素敏感性和葡萄糖利用增加相关[31]。本例突变的患者具有严重的胰岛素抵抗,提示突变与胰岛素敏感性的关系有待进一步的研究。此外,非酒精性脂肪性肝病(nonalcoholic fatty liver disease, NAFLD)常常出现在BBS患者儿童早期,可导致肝功能异常,如果不进行干预,NAFLD会随着年龄的增长而逐渐恶化。本例患者的NAFLD通过低热量饮食、二甲双胍和小檗碱治疗后取得了较好的临床疗效,因此对BBS患者的NAFLD进行早期筛查、干预和治疗十分重要。有研究发现BBS患者中亚临床甲状腺功能减退的患病率不断增加,占19.4%[8],而本例患者也表现出了早发的亚临床甲状腺功能减退症,说明BBS患者中常规进行甲状腺功能筛查的重要性。此外,在非BBS患者中,研究发现亚临床甲状腺功能减退与胰岛素抵抗、非酒精性脂肪肝、肥胖的发生发展密切相关,小剂量甲状腺素替代治疗能够改善患者的代谢情况[32~34],然而在BBS中有关亚临床甲状腺功能减退的替代治疗尚缺乏足够的研究。本例BBS患者存在轻度亚临床甲状腺功能减退,随访中促甲状腺激素水平一直稳定在5 μIU/mL左右,并且肝脏脂肪含量逐渐下降,故暂未行甲状腺素替代治疗。
BBS患者需要针对各种症状进行多学科管理和长期的评估随访。目前对于BBS患者并发症的处理主要为对症治疗,但对于一些严重的并发症比如失明,并没有有效的治疗措施。在小鼠()模型中,用于BBS视网膜色素变性的基因治疗显示出小鼠视网膜电图功能的改善趋势[35],基因治疗是BBS治疗领域的一个新进展,但具体治疗效果仍需进一步的探索和验证。本例患者的糖尿病和NAFLD的治疗取得了较好的效果,但是患者的体重控制不理想。既往研究发现在BBS小鼠模型中,下丘脑弓状核内的瘦素抵抗以及调节能量平衡的神经肽POMC生成受损等可能是BBS患者的肥胖发病机制,而黑素皮质素受体激动剂可降低小鼠的体重和食物摄入量[36~39],这对于改善BBS肥胖可能具有重要意义。有研究显示Roux-en-Y胃旁路术或袖状胃切除术可能对严重肥胖的BBS患者安全有效[40, 41],然而本文患者拒绝接受减肥手术。对BBS患者进行减肥手术的有效性和安全性有待进一步研究。
综上所述,本文报告了在一例BBS患者中发现的基因两个新突变(c.188delC, p.T63fs和c.1993_ 1995del, p.665_665del)。全外显子组测序对具有不典型症状BBS患者的确诊具有十分重要的意义。
[1] Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey., 1999, 36(6): 437–446.
[2] Suspitsin EN, Imyanitov EN. Bardet-Biedl syndrome., 2016, 7(2): 62–71.
[3] Janssen S, Ramaswami G, Davis EE, Hurd T, Airik R, Kasanuki JM, Van Der Kraak L, Allen SJ, Beales PL, Katsanis N, Otto EA, Hildebrandt F. Mutation analysis in Bardet-Biedl syndrome by DNA pooling and massively parallel resequencing in 105 individuals., 2011, 129(1): 79–90.
[4] Farag TI, Teebi AS. High incidence of Bardet Biedl syndrome among the bedouin., 1989, 36(6): 463–464.
[5] Mitchison HM, Valente EM. Motile and non-motile cilia in human pathology: from function to phenotypes., 2017, 241(2): 294–309.
[6] National Clinical Research Center for Child Health, Rare Disease Group of Chinese Pediatric Society of Chinese Medical Association. Expert consensus on the diagnosis and treatment of Bardet-Biedl syndrome in children in China., 2022, 37(4): 241–247.
国家儿童健康与疾病临床医学研究中心, 中华医学会儿科学分会罕见病学组. 中国儿童Bardet-Biedl综合征诊治专家共识. 中国实用儿科杂志, 2022, 37(4): 241–247.
[7] Álvarez-Satta M, Castro-Sánchez S, Valverde D. Bardet-Biedl syndrome as a chaperonopathy: dissecting the major role of chaperonin-like bbs proteins (BBS6-BBS10-BBS12)., 2017, 4: 55.
[8] Mujahid S, Hunt KF, Cheah YS, Forsythe E, Hazlehurst JM, Sparks K, Mohammed S, Tomlinson JW, Amiel SA, Carroll PV, Beales PL, Huda MSB, Mcgowan BM. The endocrine and metabolic characteristics of a large Bardet-Biedl syndrome clinic population., 2018, 103(5): 1834–1841.
[9] Zhai YH, Xu H, Shen Q, Wu BB. Novel mutations in the BBS12 gene of two Chinese Bardet-Biedl syndrome pedigrees., 2018, 34(8): 592–600.
翟亦晖, 徐虹, 沈茜, 吴冰冰. 儿童巴尔得–别德尔综合征2个家系研究——BBS12基因新突变. 中华肾脏病杂志, 2018, 34(8): 592–600.
[10] Schwartz R, Hildebrandt F, Benzing T, Katsanis N. Mechanisms of disease ciliopathies., 2011, 16364(21): 1533–1543.
[11] Loktev AV, Zhang QH, Beck JS, Searby CC, Scheetz TE, Bazan JF, Slusarski DC, Sheffield VC, Jackson PK, Nachury MV. A BBSome subunit links ciliogenesis, microtubule stability, and acetylation., 2008, 15(6): 854–865.
[12] Wei Q, Zhang YX, Li YJ, Zhang Q, Ling K, Hu JH. The BBSome controls IFT assembly and turnaround in cilia., 2012, 14(9): 950–957.
[13] Nachury MV, Loktev AV, Zhang QH, Westlake CJ, Peränen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, Jackson PK. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis., 2007, 129(6): 1201–1213.
[14] Billingsley G, Bin J, Fieggen KJ, Duncan JL, Gerth C, Ogata K, Wodak SS, Traboulsi EI, Fishman GA, Paterson A, Chitayat D, Knueppel T, Millán JM, Mitchell GA, Deveault C, Héon E. Mutations in chaperonin-like BBS genes are a major contributor to disease development in a multiethnic Bardet-Biedl syndrome patient population., 2010, 47(7): 453–463.
[15] Cardenas-Rodriguez M, Badano JL. Ciliary biology: understanding the cellular and genetic basis of human ciliopathies., 2009, 151C(4): 263–280.
[16] Pawlik B, Mir A, Iqbal H, Li Y, Nürnberg G, Becker C, Qamar R, Nürnberg P, Wollnik B. A novel familialBBS12 mutation associated with a mild phenotype: implications for clinical and molecular diagnostic strategies., 2010, 1(1): 27–34.
[17] Khan SA, Muhammad N, Khan MA, Kamal A, Rehman ZU, Khan S. Genetics of human Bardet-Biedl syndrome, an updates., 2016, 90(1): 3–15.
[18] Stoetzel C, Muller J, Laurier V, Davis EE, Zaghloul NA, Vicaire S, Jacquelin C, Plewniak F, Leitch CC, Sarda P, Hamel C, De Ravel TJL, Lewis RA, Friederich E, Thibault C, Danse JM, Verloes A, Bonneau D, Katsanis N, Poch O, Mandel JL, Dollfus H. Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome., 2007, 80(1): 1–11.
[19] Nikkhah E, Safaralizadeh R, Mohammadiasl J, Tahmasebi Birgani M, Hosseinpour Feizi MA, Golchin N. Identification of a novel compound heterozygous mutation in BBS12 in an iranian family with Bardet-Biedl syndrome using targeted next generation sequencing., 2018, 20(2): 284–289.
[20] Castro-Sánchez S, Álvarez-Satta M, Cortón M, Guillén E, Ayuso C, Valverde D. Exploring genotype-phenotype relationships in Bardet-Biedl syndrome families., 2015, 52(8): 503–513.
[21] Deveault C, Billingsley G, Duncan JL, Bin J, Theal R, Vincent A, Fieggen KJ, Gerth C, Noordeh N, Traboulsi EI, Fishman GA, Chitayat D, Knueppel T, Millán JM, Munier FL, Kennedy D, Jacobson SG, Innes AM, Mitchell GA, Boycott K, Héon E. BBS genotype-phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition., 2011, 32(6): 610–619.
[22] Baker K, Beales PL. Making sense of cilia in disease: the human cilioplathies., 2009, 151C(4): 281–295.
[23] Hamel CP. Cone rod dystrophies., 2007, 2: 7.
[24] Adams NA, Awadein A, Toma HS. The retinal ciliopathies., 2007, 28(3): 113–125.
[25] Pomeroy J, Krentz AD, Richardson JG, Berg RL, Vanwormer JJ, Haws RM. Bardet-Biedl syndrome: weight patterns and genetics in a rare obesity syndrome., 2021, 16(2): e12703.
[26] Forsythe E, Sparks K, Best S, Borrows S, Hoskins B, Sabir A, Barrett T, Williams D, Mohammed S, Goldsmith D, Milford DV, Bockenhauer D, Foggensteiner L, Bealest PL. Risk factors for severe renal disease in Bardet-Biedl syndrome., 2017, 28(3): 963–970.
[27] Harnett JD, Green JS, Cramer BC, Johnson G, Chafe L, Mcmanamon P, Farid NR, Pryse-Phillips W, Parfrey PS. The spectrum of renal disease in Laurence-Moon-Biedl syndrome., 1988, 319(10): 615–618.
[28] Imhoff O, Marion V, Stoetzel C, Durand M, Holder M, Sigaudy S, Sarda P, Hamel CP, Brandt C, Dollfus H, Moulin B. Bardet-Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort., 2011, 6(1): 22–29.
[29] Starks RD, Beyer AM, Guo DF, Boland L, Zhang QH, Sheffield VC, Rahmouni K. Regulation of insulin receptor trafficking by Bardet Biedl syndrome proteins., 2015, 11(6): e1005311.
[30] Gerdes JM, Christou-Savina S, Xiong Y, Moede T, Moruzzi N, Karlsson-Edlund P, Leibiger B, Leibiger IB, Östenson CG, Beales PL, Berggren PO. Ciliary dysfunction impairs beta-cell insulin secretion and promotes development of type 2 diabetes in rodents., 2014, 5: 5308.
[31] Marion V, Mockel A, De Melo C, Obringer C, Claussmann A, Simon A, Messaddeq N, Durand M, Dupuis L, Loeffler J-P, King P, Mutter-Schmidt C, Petrovsky N, Stoetzel C, Dollfus H. BBS-induced ciliary defect enhances adipogenesis, causing paradoxical higher-insulin sensitivity, glucose usage, and decreased inflammatory response., 2012, 16(3): 363–377.
[32] Maratou E, Hadjidakis DJ, Kollias A, Tsegka K, Peppa M, Alevizaki M, Mitrou P, Lambadiari V, Boutati E, Nikzas D, Tountas N, Economopoulos T, Raptis SA, Dimitriadis G. Studies of insulin resistance in patients with clinical and subclinical hypothyroidism., 2009, 160(5): 785–790.
[33] Walczak K, Sieminska L. Obesity and thyroid axis., 2021, 18(18): 9434.
[34] Bruinstroop E, Dalan R, Cao Y, Bee YM, Chandran K, Cho LW, Soh SB, Teo EK, Toh SA, Leow MKS, Sinha RA, Sadananthan SA, Michael N, Stapleton HM, Leung C, Angus PW, Patel SK, Burrell LM, Lim SC, Sum CF, Velan SS, Yen PM. Low-dose levothyroxine reduces intrahepatic lipid content in patients with type 2 diabetes mellitus and NAFLD., 2018, 103(7): 2698–2706.
[35] Seo S, Mullins RF, Dumitrescu AV, Bhattarai S, Gratie D, Wang K, Stone EM, Sheffield V, Drack AV. Subretinal gene therapy of mice with Bardet-Biedl syndrome type 1., 2013, 54(9): 6118–6132.
[36] Rahmouni K, Fath MA, Seo S, Thedens DR, Berry CJ, Weiss R, Nishimura DY, Sheffield VC. Leptin resistance contributes to obesity and hypertension in mouse models of Bardet-Biedl syndrome., 2008, 118(4): 1458–1467.
[37] Seo S, Guo DF, Bugge K, Morgan DA, Rahmouni K, Sheffield VC. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling., 2009, 18(7): 1323–1331.
[38] Oh EC, Vasanth S, Katsanis N. Metabolic regulation and energy homeostasis through the primary cilium., 2015, 21(1): 21–31.
[39] Wang LH, Liu Y, Stratigopoulos G, Panigrahi S, Sui LN, Zhang YY, Leduc CA, Glover HJ, De Rosa MC, Burnett LC, Williams DJ, Shang LS, Goland R, Tsang SH, Wardlaw S, Egli D, Zheng D, Doege CA, Leibel RL. Bardet-Biedl syndrome proteins regulate intracellular signaling and neuronal function in patient-specific iPSC- derived neurons., 2021, 131(8): e146287.
[40] Daskalakis M, Till H, Kiess W, Weiner RA. Roux-en-Y gastric bypass in an adolescent patient with Bardet-Biedl syndrome, a monogenic obesity disorder., 2010, 20(1): 121–125.
[41] Boscolo M, Féry F, Cnop M. Beneficial outcomes of sleeve gastrectomy in a morbidly obese patient with Bardet-Biedl syndrome., 2017, 1(4): 317–322.
Diagnosis and genetic analysis of a case with Bardet-Biedl syndrome caused by compound heterozygous mutations in thegene
Yanting Shen1,2, Yan Ling1,2, Zhiqiang Lu1,2, Xiaomu Li1,2, Hua Bian1,2, Hongmei Yan1,2, Mingfeng Xia1,2, Xinxia Chang1,2, Jingjing Jiang1,2, Jing Zhang1,2, Xin Gao1,2
Bardet-Biedl syndrome (BBS) is a rare autosomal recessive ciliopathy, which is caused by mutations mainly in genes encoding BBSome complex and IFT complex. Here, we reported a 21-year-old female with BBS characterized by three primary features including obesity, retinitis pigmentosa sine pigmento and bilateral renal cysts. She also had some secondary features such as diabetes mellitus, nonalcoholic fatty liver disease, subclinical hypothyroidism and mild conductive hearing damage. Whole exome sequencing revealed two compound heterozygous mutations in exon 2 of thegene (c.188delC, p.T63fs and c.1993_1995del, p.665_665del) in this patient. Sanger sequencing showed that her father and mother carried c.188delC (p.T63fs) and c.1993_1995del (p.665_665del) variants, respectively, while her parents were free of BBS-related symptoms. In conclusion, this case reported two novel mutations (c.188delC, p.T63fs and c.1993_1995del, p.665_665del) of thegene in a girl presented with BBS, which provides novel genetic resources for studies of the disease. Meanwhile, the BBS case shows the entire development progress from her birth to adulthood, which helps facilitate clinicians’ understanding of BBS.
Bardet-Biedl syndrome;gene; whole exome sequencing
2022-05-30;
2022-08-26;
2022-09-08
国家自然科学基金项目(编号:81770865)资助[Supported by the National Natural Science Foundation of China (No. 81770865)]
沈艳婷,在读硕士研究生,专业方向:内分泌与代谢病。E-mail: 19211210076@fudan.edu.cn
张晶,博士,研究方向:内分泌与代谢病。E-mail: zhang_jing2016@126.com
10.16288/j.yczz.22-182
(责任编委: 周红文)
猜你喜欢
湖北农业科学(2022年11期)2022-07-18
心电与循环(2021年4期)2021-11-29
锻压装备与制造技术(2020年6期)2021-01-25
中国生殖健康(2020年4期)2021-01-18
实用肿瘤学杂志(2020年4期)2020-12-08
宝藏(2020年3期)2020-10-14
湖北农业科学(2014年11期)2014-09-10
医学综述(2011年12期)2011-12-09
亚热带农业研究(2011年3期)2011-09-29
职业·中旬(2009年12期)2009-06-01
