
LMNA基因突变相关脂肪萎缩综合征的研究进展
2022-11-21 03:19:14肖诚刘洁颖杨春如于淼
遗传 2022年10期
肖诚,刘洁颖,2,杨春如,于淼
综 述
基因突变相关脂肪萎缩综合征的研究进展
肖诚1,刘洁颖1,2,杨春如1,于淼1
1. 中国医学科学院北京协和医学院,北京协和医院内分泌科,国家卫生健康委员会内分泌重点实验室,北京 100730 2. 中国医学科学院北京协和医学院,北京协和医院医学科学研究中心,疑难重症及罕见病国家重点实验室,北京 100730
基因突变相关脂肪萎缩综合征(lipodystrophy syndrome)是一组由A型核纤层蛋白(lamin A/C,)基因突变引起的常染色体显性遗传单基因疾病,以选择性脂肪缺失伴胰岛素抵抗等代谢异常为特征。本文总结了目前已报道的可引起脂肪萎缩综合征的突变位点,及该突变位点导致的代谢并发症、心血管异常、性腺轴紊乱、肌病、肾脏异常等多种临床表现,阐述了基因致病性突变位点可能的致病机制及诊疗方法,以期为该疾病的基础研究和临床诊治提供参考。
脂肪萎缩综合征;基因突变;胰岛素抵抗;代谢紊乱性疾病;致病机制
脂肪萎缩综合征(lipodystrophy syndrome)是一组以脂肪选择性不同程度丢失为特征的异质性疾病,发病率约为1/1,000,000[1,2]。A型核纤层蛋白(lamin A/C,)基因突变相关脂肪萎缩综合征包含全身性或部分性脂肪萎缩。其中部分性脂肪萎缩又名为家族性部分性脂肪萎缩综合征型2型(familial partial lipodystrophy,FPLD2,OMIM#1516620),该病多在青春期发病,典型表现为四肢和躯干皮下脂肪减少,内脏和面颈部区域脂肪堆积。突变相关全身性脂肪萎缩则表现为全身脂肪减少,常与突变相关早衰综合征重叠[1,3]。突变相关的脂肪萎缩综合征可出现多种临床表型,累及全身多系统,包括胰岛素抵抗、糖尿病、重度高甘油三酯血症、多囊卵巢综合征(polycystic ovary syndrome,PCOS)等多种内分泌代谢紊乱疾病,以及心血管、肾脏损害等[4]。
迄今为止至少存在500余种突变形式,可引起包括FPLD2在内的十余种疾病,累及脂肪、心肌、骨骼肌、骨骼、皮肤、神经等多种组织,统称为核纤层蛋白病[5]。所编码的A型核纤层蛋白(lamin A/C)是核膜内侧核纤层的重要组成蛋白,在维持细胞核结构和调节转录因子方面发挥重要作用[6]。然而,突变相关脂肪萎缩的具体发病机制尚未完全阐明,目前临床治疗多为对症[4,7,8]。对于脂肪萎缩综合征来说,早期正确诊断至关重要,本文通过总结不同位点突变相关脂肪萎缩综合征的临床表现、主要致病机制和诊疗方法,以期为该病的基础研究和临床诊治提供更多的遗传学依据,最大程度地延缓疾病进展,改善预后。
1 LMNA基因突变相关脂肪萎缩综合征的致病基因与表型的关联
基因突变相关脂肪萎缩综合征可以出现多种临床表现,其中糖脂代谢紊乱和心血管系统异常是最为常见的临床表现,此外还包括性腺轴紊乱、肌病、肾脏病变以及皮肤病变等临床表现。研究发现80%的FPLD2由第482位密码子突变引起,如p.R482W,形成典型的FPLD2[9]。其他突变散布于整个基因,大多为错义突变,无义突变、重复和剪接突变均有报道(http://www.umd.be/LMNA/)。FPLD2主要是由外显子8和外显子11发生的杂合或复合杂合突变引起[10]。
Lamin A/C主要由3个部分组成:氨基端头部、羧基端尾部以及一个α螺旋的杆状结构域(包含C1a、L1、C1b、L12、C2区域)[11]。本文主要总结了既往研究中除482位密码子外其他可引起脂肪萎缩综合征的突变位点(图1),及各突变位点所在区域出现的突变比例(表1)。进一步归纳了相应的临床表现,包括代谢并发症、心血管异常表现、性腺轴紊乱、肌病、肾脏异常等(表2),大部分致病突变均为杂合突变,少部分为纯合突变。表3总结了表2中所有突变位点引起的临床表现发生率,其中高脂血症的发生率高达91.4%,糖尿病的发生率为88.5%,45.7%的突变位点会出现胰岛素抵抗,性腺轴紊乱和肝脏脂肪变性的发生率分别为42.3%和42.9%,心脏相关的病变包括心脏传导异常、心律失常、心肌病变、瓣膜病变、心力衰竭,发生率分别占11.4%,25.7%,31.4%,26.9%和17.1%。此外,肌病、肾脏病变和皮肤病变的分别占25.8%、22.9%和28.6%。
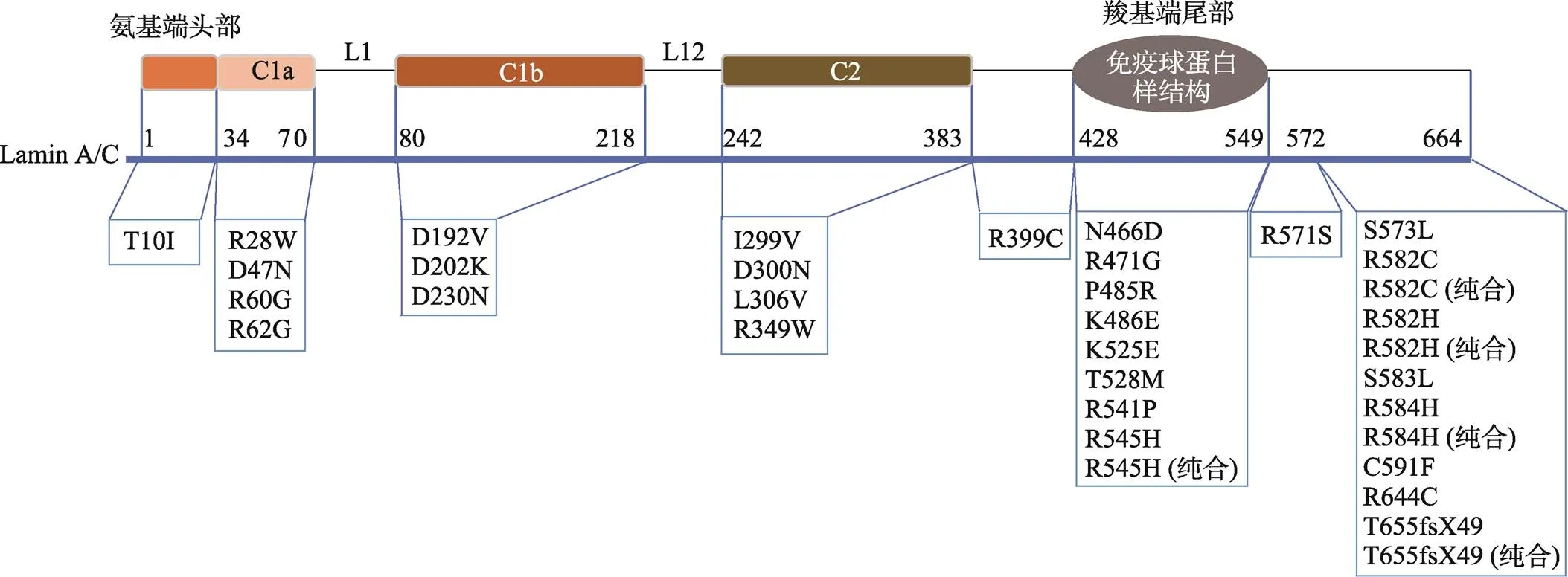
图1 LMNA突变相关脂肪萎缩综合征致病位点所在区域

表1 LMNA突变相关脂肪萎缩综合征致病位点所在区域的比例
1.1 糖脂代谢紊乱性疾病
脂肪萎缩综合征容易出现以胰岛素抵抗为核心包括高血糖、高脂血症、非酒精性脂肪肝病等在内的多种代谢并发症。一项源自土耳其关于FPLD2的研究显示,与对照组相比,FPLD2患者的甘油三酯(triglyceride,TG)及血糖水平明显升高,而高密度脂蛋白水平下降[24]。由于脂肪减少会突出肌肉外观,因此相比男性而言,女性患者更容易辨别,其代谢并发症也更严重。一项纳入258例FPLD2患者的研究发现大多数男性患者TG水平为400 mg/dL或更低,而女性患者的TG平均水平达1000~2000 mg/dL,容易罹患急性胰腺炎等并发症[61]。
1.2 心血管疾病异常
在脂肪萎缩综合征中,心肌、节律传导系统、心脏瓣膜和冠状动脉等均可出现异常表现。携带突变的人群心律失常或心肌病变每年的发病率为8.43/1000人,高于未携带人群的6.38/1000人[62]。心肌病变通常表现为扩张型心肌病、心脏传导阻滞(Ⅰ度、Ⅱ度和Ⅲ度)、室上性心律失常(房扑、房颤)、室性心率失常(室颤、持续室速)、心动过缓-过速综合征[63]、心力衰竭、左室心尖部室壁瘤等[64],甚至出现猝死[65,19]。无干预的情况下,突变引起的心脏受累患者通常预后不佳。Meta分析显示携带突变的人群猝死风险较普通人群升高3.7倍[66]。一项随访7年纳入122名携带突变患者的研究更是证实了这些患者存在恶性室性心律失常的倾向,34%的患者出现持续性室性心律失常,48%的患者需要一级预防心源性猝死装用植入式心脏复律除颤器[65]。当突变的心肌病变患者在左室射血分数≤45%或起搏百分比≥50%时,心脏再同步化治疗有效[67]。
1.3 骨骼肌异常表现
突变可导致先天性肌营养不良、腓骨肌萎缩症2B1型、肢带型肌营养不良1B、常染色体显性遗传Emery-Dreifuss 型肌营养不良等,表现为颈轴肌、肩腓肌无力和肌萎缩,关节挛缩,脊柱畸形等异常[68]。此外,研究发现FPLD2患者的肌肉更加发达,肌肉活检可发现1型和2型肌纤维肥大,以及出现一些非特异性改变[69]。在一些脂肪萎缩的患者中也会出现多种骨骼肌异常的表现。例如,一项关于p.R349W突变家系报道中,多名患者出现了肌痛以及脊柱侧弯等病变[24]。p.R471G突变则可以引起肌无力和关节挛缩等异常表现[39]。p.R644C突变的患者则出现了肌痛、肌无力、肌萎缩、脊柱病变等几乎所有骨骼肌病变的表现[57,58]。
1.4 性腺轴紊乱
不少研究报道了FPLD2患者存在性腺轴紊乱,以PCOS常见,少部分患者也可以出现月经稀发[20]、高促性腺激素性腺功能减退症[70]。在FPLD2中,除了与胰岛素抵抗相关,PCOS可能还与游离脂肪酸积聚引起的卵巢脂毒性以及腹腔内脂肪组织的堆积相关[71]。
1.5 肾脏异常表现
p.R349W突变的FPLD2家系中共有12名患者携带该突变,4名患者表现出不同程度的肾功能不全和蛋白尿,肾脏活检病理显示为局灶节段性肾小球硬化症[72]。p.R482W突变的FPLD2患者肾脏活检病理为2型系膜毛细血管增生性肾小球肾炎[73]。此外,存在糖尿病时还可引起糖尿病肾病的并发症[34]。
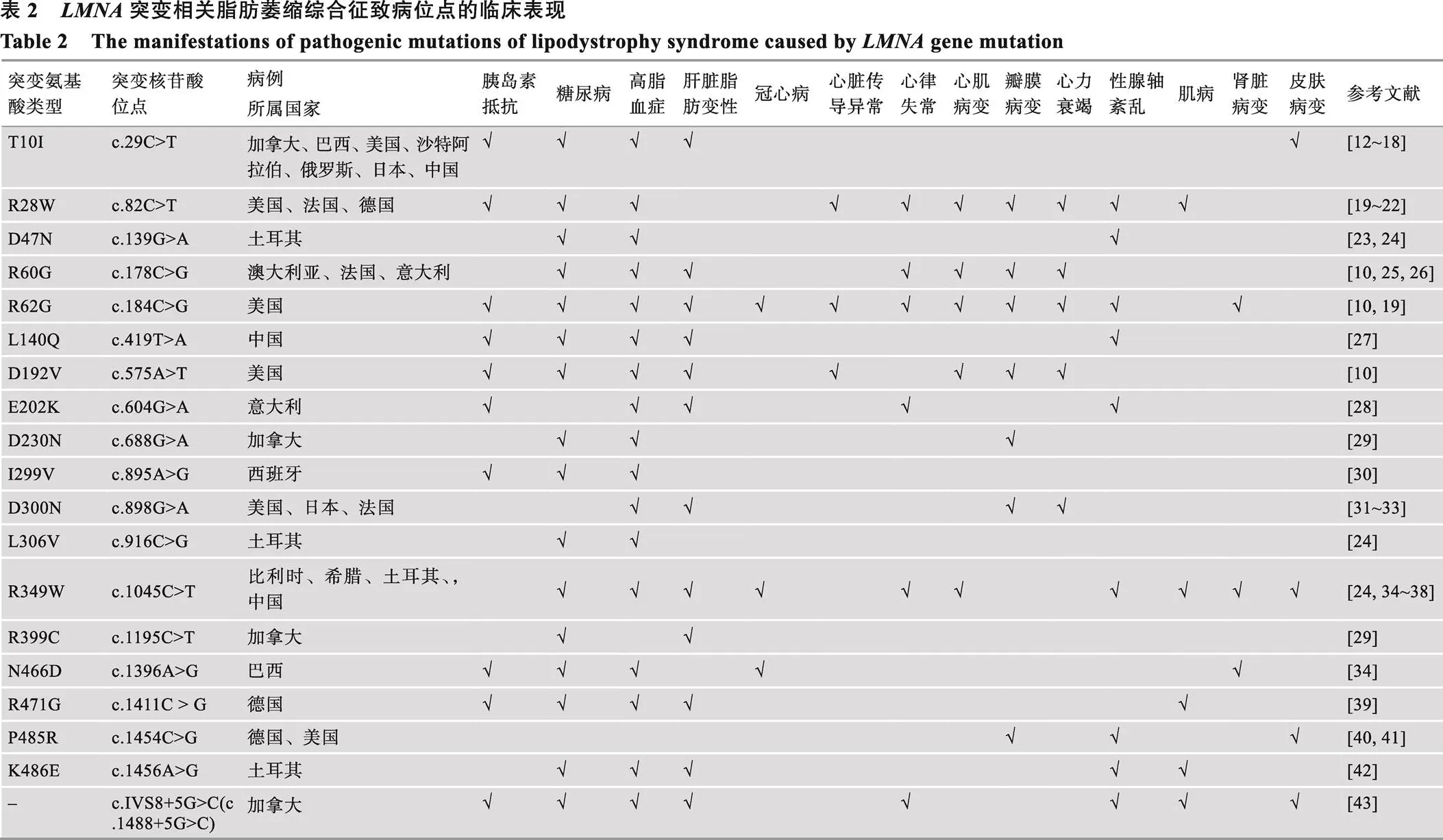

表3 LMNA突变相关脂肪萎缩综合征的临床表现的比例
1.6 其他表现
一些较为少见的临床表现包括如皮肤萎缩、硬化、黄瘤、天鹅绒浅棕色乳头瘤增生性斑块、皮赘等异常表现[12,40,43,48~50,53];脐疝[42,49,50,52]、智力异常[39,48,50]、听力下降[24,34,57, 58]在FPLD2患者中亦有报道。
2 LMNA基因突变相关脂肪萎缩综合征的致病机制
关于突变产生组织特异性的表型,相同的突变位点可引起不同的疾病表型的具体机制尚不明确,但是根据目前已有的研究报道,主要致病机制大致分为以下三类[74](图2)。
2.1 错误加工或未加工的核纤层蛋白A前体(prelamin A)积聚,产生细胞毒性
基因包含12个外显子,经过转录翻译形成prelamin A,肽链的羧基端“CAAX”(C,半胱氨酸;A,脂肪族氨基酸;X,末端氨基酸)结构域的半胱氨酸被法尼基转移酶识别发生法尼基化,半胱氨酸再发生甲基酯化,然后由蛋白酶ZMPSTE24剪切掉包含“AAX”在内的末端15个氨基酸,形成不含法尼基化残基的成熟Lamin A[75,76]。突变后阻碍了prelamin A的成熟过程,法尼基化的prelamin A锚定在核膜上,容易导致核膜结构紊乱,功能异常[77,78]。在一项研究中发现与对照组细胞相比,携带p.D47Y、p. L92F、p.L387V、p.R399H、p.L421P突变的成纤维细胞核形状异常、增殖活性降低,细胞中prelamin A积聚,氧化应激水平增加,线粒体呼吸链蛋白表达减少,细胞亦过早衰老。抑制prelamin A的法尼基化可以防止氧化应激和细胞衰老[79]。此外,他汀类药物或抗氧化剂预处理可以部分改善携带p.R482W突变的内皮细胞功能[80]。
2.2 Lamin A/C与染色质或蛋白之间的相互作用
2.2.1 减少胆固醇调节元件结合蛋白1(sterol regulatory element binding protein 1,SREBP1)表达
SREBP1是一种脂肪细胞转录因子,其激活对于启动间充质干细胞分化为脂肪细胞的至关重要。Lloyd等[81]首次鉴定SREBP1是Lamin A的一种新型互作因子,p. G465D、p.R482W和p.K486N显著降低Lamin A与SREBP1的结合,减少了SREBP1表达,是引起代谢紊乱的原因之一。
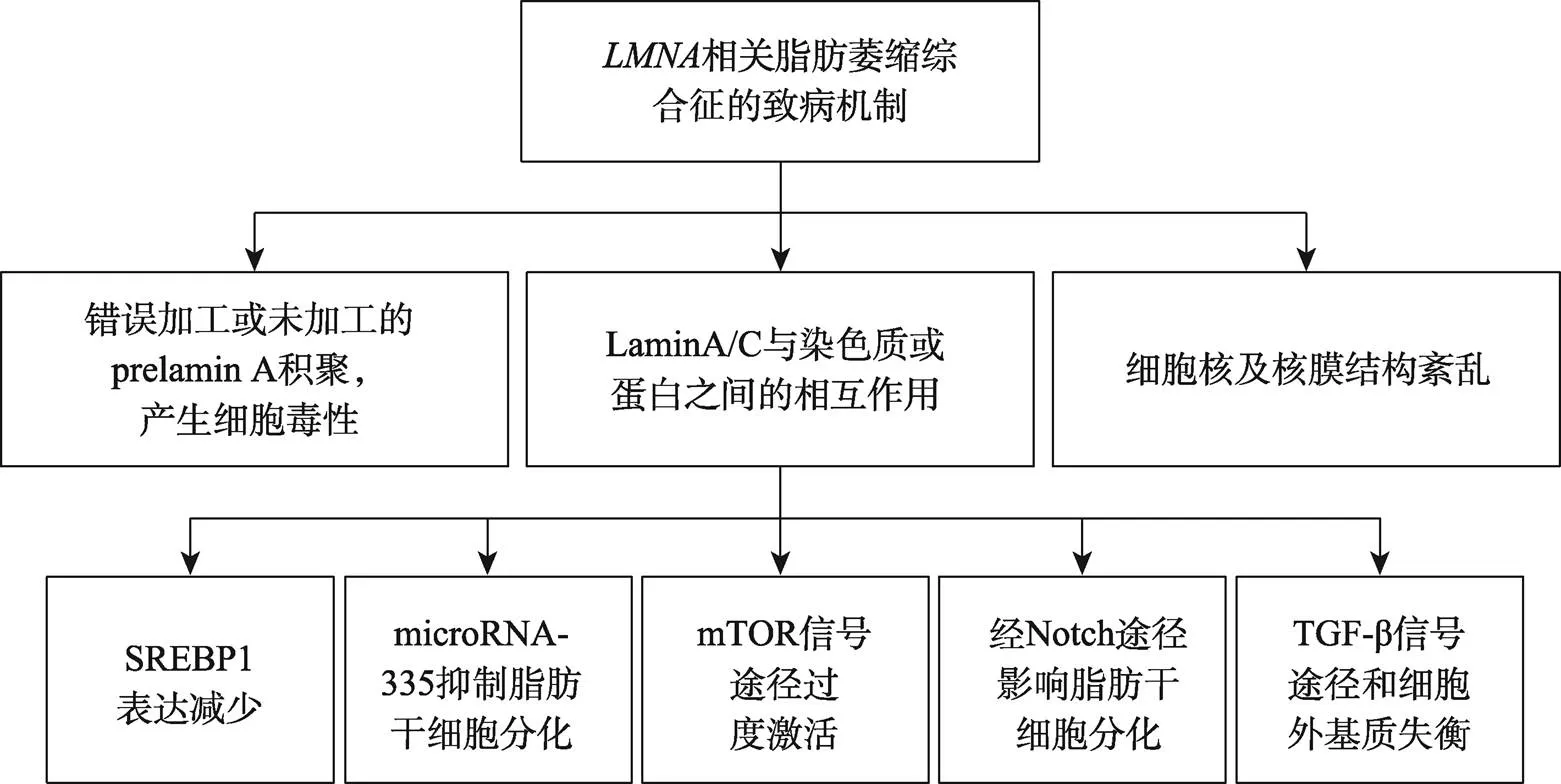
图2 LMNA突变相关脂肪萎缩综合征的致病机制
2.2.2 miR-335抑制脂肪干细胞分化
microRNA是短链非编码 RNA,通常在与3′非翻译区结合后通过降解或翻译沉默来下调靶mRNA表达。miR-335促进成肌,抑制间充质干细胞向脂肪细胞和骨细胞分化[82,83]。在野生型的脂肪干细胞成脂分化过程中,结合miR-335基因座,miR-335表达受抑制,而携带p.R482W突变的脂肪干细胞和源于FPLD2患者的成纤维细胞中miR-335表达升高,突变后失去了与miR-335的结合,从而使其发生持续转录,抑制脂肪干细胞分化[84]。
2.2.3哺乳动物雷帕霉素靶标(mammalian target of rapamycin,mTOR)信号途径
mTOR信号通路在调节脂肪细胞分化、脂质代谢、产热和脂肪因子合成与分泌中发挥关键作用。H222P/H222P小鼠拟似突变引起的骨骼肌、心肌异常表型,在该小鼠模型中应用mTOR的抑制剂西罗莫司,激活了自噬水平,减缓了心功能减退[85]。–/–小鼠模型的脂肪减少与脂肪分解有关,雷帕霉素治疗后脂肪分解减少,有助于改善全身代谢[86]。
2.2.4 Notch信号途径
脂肪组织过表达Notch细胞内结构域(Notch intracellular domain, NICD)的C57BL/6J小鼠呈现多种代谢异常表现,且小鼠的白色脂肪含量明显减低,出现异位脂肪沉积,呈现出类似脂肪萎缩的表型[87]。在人心脏间充质干细胞中表达野生型或p.R482L,进一步过表达NICD,相比野生组,突变组脂肪细胞分化能力下降,突变可能通过Notch信号通路影响脂肪细胞分化[88]。
2.2.5 转化生长因子β(transforming growth factor- β, TGF-β)途径
FPLD2患者的脂肪组织中TGF-β和细胞外基质的失衡也是致病原因之一。与FPLD2患者类似,脂肪组织特异性表达p.R482Q突变小鼠的脂肪组织纤维化程度增加,细胞外基质发生重塑,归因于TGF-β和基质金属蛋白酶表达增加[89]。
2.3 细胞核及核膜结构紊乱
几乎所有的突变都会出现核形态的异常和核膜结构的紊乱,如细胞核增大,核膜出现起泡、凹陷以及异常积聚,削弱了对外部刺激的抵抗力,也可能会改变与核纤层关联域(Lamina associated domains,LADs)相关蛋白或DNA,进而影响染色质结构[90,91]。
3 诊断和治疗
遗传分析有助于建立基因突变相关脂肪萎缩综合征的诊断并指导进一步治疗。治疗上尚无针对性和使脂肪组织再生的方法,主要是预防或改善脂肪萎缩综合征的并发症。
3.1 基础治疗方法:饮食和运动
由于脂肪萎缩综合征存在多种代谢相关的并发症,因此正确的生活方式是治疗的基石,鼓励大多数患者遵循均衡饮食,在没有特定禁忌症的情况下进行运动[4,92]。
3.2 瘦素疗法
外源性补充瘦素,可能有助于改善FPLD2的代谢情况。一项含22例FPLD2患者的前瞻性研究显示瘦素类似物美曲普汀改善了TG和糖化血红蛋白[7]。在中至重度的低瘦素血症FPLD2患者中应用美曲普汀,TG以及胰岛素敏感性和分泌指数也得到了有效改善[93]。
3.3 美容治疗
通过接受自体脂肪组织移植或真皮填充剂的植入,可以改善面部或乳房外观。头部、颈部或外阴的多余脂肪可以通过手术减少或通过吸脂术减少。
3.4 针对合并症的治疗
3.4.1 糖尿病
二甲双胍是一线降糖和改善胰岛素抵抗用药。噻唑烷二酮类药物可能有助于改善代谢并发症[1]。胰岛素可有效降低高血糖,但可能需要极大剂量胰岛素[4]。新型降糖药物如胰高糖素样肽-1受体激动剂、二肽基肽酶4抑制剂和钠-葡萄糖协同转运蛋白-2抑制剂,均能够有效降低多种心血管危险因素,但这些药物尚缺乏在脂肪萎缩综合征中系统性的研究[92]。Roux-en-Y胃旁路代谢手术有助于减轻体重及改善代谢[94]。
3.4.2 高脂血症
他汀类药物应作为一线用药。对于重度高TG的患者,可考虑贝特类药物。Omega-3-脂肪酸也被广泛使用,但这一特定人群中的功效缺乏充足的证据[1]。其他新的血脂异常药物如前蛋白转化酶枯草杆菌蛋白酶9型抑制剂尚未在脂肪萎缩的患者中进行研究。
3.4.3 非酒精性脂肪性肝病
在饮食和运动方法的基础上,噻唑烷二酮类药物和维生素E活性有助于改善肝功能或肝脂肪变性[95,96],但这些药物缺乏在脂肪萎缩综合征患者中的研究。
4 结语与展望
突变相关脂肪萎缩综合征的致病位点分布于整个区域,突变类型多变,致病机制复杂,涉及多种信号通路,但该病特异性的分子机制仍不清晰,需要进一步研究来揭示不同突变位点的特异性致病机制。该病临床表现多样,异质性较高,在临床工作中容易漏诊、误诊,贻误病情,影响预后。本文通过总结突变相关脂肪萎缩综合征的致病位点相关临床表现以及可能的致病机制,有助于提高临床医生中对该疾病的认识,有利于早期诊断和管理,最大程度改善患者预后。也为将来深入研究突变引起的核纤层蛋白病奠定了基础。随着研究的深入,相信未来将更深层次地认识突变相关脂肪萎缩综合征并为患者提供更好的治疗措施。
[1] Garg, A. Clinical review#: lipodystrophies: genetic and acquired body fat disorders, 2011, 96(11): 3313–3325.
[2] Garg A. Lipodystrophies, 2000, 108(2): 143– 152.
[3] Mann JP, Savage DB. What lipodystrophies teach us about the metabolic syndrome, 2019, 129(10): 4009–4021.
[4] Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, Mungai L, Oral EA, Patni N, Rother KI, von Schnurbein J, Sorkina E, Stanley T, Vigouroux C, Wabitsch M, Williams R, Yorifuji T, The diagnosis and management of lipodystrophy syndromes: a multi-society practice guideline, 2016, 101(12): 4500–4511.
[5] Schreiber KH, Kennedy BK. When lamins go bad: nuclear structure and disease, 2013, 152(6): 1365–1375.
[6] Osmanagic-Myers S, Foisner R. The structural and gene expression hypotheses in laminopathic diseases-not so different after all, 2019, 30(15): 1786– 1790.
[7] Sekizkardes H, Cochran E, Malandrino N, Garg A, Brown RJ. Efficacy of metreleptin treatment in familial partial lipodystrophy due to PPARG vspathogenic variants, 2019, 104(8): 3068– 3076.
[8] Diker-Cohen T, Cochran E, Gorden P, Brown RJ. Partial and generalized lipodystrophy: comparison of baseline characteristics and response to metreleptin, 2015, 100(5): 1802–1810.
[9] Bertrand AT, Chikhaoui K, Yaou RB, Bonne G. Clinical and genetic heterogeneity in laminopathies, 2011, 39(6): 1687–1692.
[10] Subramanyam L, Simha V, Garg A. Overlapping syndrome with familial partial lipodystrophy, Dunnigan variety and cardiomyopathy due to amino-terminal heterozygous missense lamin A/C mutations, 2010, 78(1): 66–73.
[11] Strelkov SV, Schumacher J, Burkhard P, Aebi U, Herrmann H. Crystal structure of the human lamin A coil 2B dimer: implications for the head-to-tail association of nuclear lamins, 2004, 343(4): 1067–1080.
[12] Mory PB, Crispim F, Kasamatsu T, Gabbay MA, Dib SA, Moisés RS. Atypical generalized lipoatrophy and severe insulin resistance due to a heterozygousp.T10I mutation, 2008, 52(8): 1252–1256.
[13] Jiajue R, Feng K, Wang R, Xia W. Recurrent femoral fractures in a boy with an atypical progeroid syndrome: a case report, 2020, 106(3): 325–330.
[14] Fukaishi T, Minami I, Masuda S, Miyachi Y, Tsujimoto K, Izumiyama H, Hashimoto K, Yoshida M, Takahashi S, Kashimada K, Morio T, Kosaki K, Maezawa Y, Yokote K, Yoshimoto T, Yamada T. A case of generalized lipodystrophy-associated progeroid syndrome treated by leptin replacement with short and long-term monitoring of the metabolic and endocrine profiles, 2020, 67(2): 211–218.
[15] Sahinoz M, Khairi S, Cuttitta A, Brady GF, Rupani A, Meral R, Tayeh MK, Thomas P, Riebschleger M, Camelo-Piragua S, Innis JW, Bishr Omary M, Michele DE, Oral EA. Potential association of-associated generalized lipodystrophy with juvenile dermatomyositis, 2018, 4: 6.
[16] Hussain I, Patni N, Ueda M, Sorkina E, Valerio CM, Cochran E, Brown RJ, Peeden J, Tikhonovich Y, Tiulpakov A, Stender SRS, Klouda E, Tayeh MK, Innis JW, Meyer A, Lal P, Godoy-Matos AF, Teles MG, Adams-Huet B, Rader DJ, Hegele RA, Oral EA, Garg A. A novel generalized lipodystrophy-associated progeroid syndrome due to recurrent heterozygousp.T10I mutation, 2018, 103(3): 1005–1014.
[17] Cardona-Hernández R, Suárez-Ortega L, Torres M. Difficult to manage diabetes mellitus associated with generalized congenital lipodystrophy. Report of two cases., 2011, 74(2): 126–130.
[18] Csoka AB, Cao H, Sammak PJ, Constantinescu D, Schatten GP, Hegele RA. Novel lamin A/C gene () mutations in atypical progeroid syndromes, 2004, 41(4): 304–308.
[19] Garg A, Speckman RA, Bowcock AM. Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/C gene, 2002, 112(7): 549–555.
[20] Decaudain A, Vantyghem MC, Guerci B, Hécart AC, Auclair M, Reznik Y, Narbonne H, Ducluzeau PH, Donadille B, Lebbé C, Béréziat V, Capeau J, Lascols O, Vigouroux C. New metabolic phenotypes in laminopathies:mutations in patients with severe metabolic syndrome, 2007, 92(12): 4835– 4844.
[21] Türk M, Wehnert M, Schröder R, Chevessier F., Multisystem disorder and limb girdle muscular dystrophy caused byp.R28W mutation, 2013, 23(7): 587–590.
[22] Ambonville C, Bouldouyre MA, Laforêt P, Richard P, Benveniste O, Vigouroux C. A complex case of diabetes due to LMNA mutation., 2017, 38(10): 695–699.
[23] Kutbay NO, Yurekli BS, Onay H, Altay CT, Atik T, Hekimsoy Z, Saygili F, Akinci B. A case of familial partial lipodystrophy caused by a novel lamin A/C () mutation in exon 1 (D47N), 2016, 29: 37–39.
[24] Akinci B, Onay H, Demir T, Savas-Erdeve Ş, Gen R, Simsir IY, Keskin FE, Erturk MS, Uzum AK, Yaylali GF, Ozdemir NK, Atik T, Ozen S, Yurekli BS, Apaydin T, Altay C, Akinci G, Demir L, Comlekci A, Secil M, Oral EA. Clinical presentations, metabolic abnormalities and end-organ complications in patients with familial partial lipodystrophy, 2017, 72: 109–119.
[25] van der Kooi AJ, Bonne G, Eymard B, Duboc D, Talim B, Van der Valk M, Reiss P, Richard P, Demay L, Merlini L, Schwartz K, Busch HF, de Visser M. Lamin A/C mutations with lipodystrophy, cardiac abnormalities, and muscular dystrophy, 2002, 59(4): 620–623.
[26] Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet Jr HJ, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Müehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease, 1999, 341(23): 1715–1724.
[27] He GY, Yan Z, Sun L, Lv Y, Guo W, Gang XK, Wang G. Diabetes mellitus coexisted with progeria: a case report of atypical Werner syndrome with novelmutations and literature review, 2019, 66(11): 961–969.
[28] Cecchetti C, D'Apice MR, Morini E, Novelli G, Pizzi C, Pagotto U, Gambineri A. Case report: an atypical form of familial partial lipodystrophy type 2 due to mutation in the rod domain of lamin A/C, 2021, 12: 675096.
[29] Lanktree M, Cao H, Rabkin SW, Hanna A, Hegele RA., Novelmutations seen in patients with familial partial lipodystrophy subtype 2 (FPLD2; MIM 151660), 2007, 71(2): 183–186.
[30] Araújo-Vilar D, Victoria B, González-Méndez B, Barreiro F, Fernández-Rodríguez B, Cereijo R, Gallego-Escuredo JM, Villarroya F, Pañeda-Menéndez A. Histological and molecular features of lipomatous and nonlipomatous adipose tissue in familial partial lipodystrophy caused bymutations, 2012, 76(6): 816–824.
[31] Mahdi L, Kahn A, Dhamija R, Vargas HE. Hepatic steatosis resulting from-associated familial lipodystrophy, 2020, 7(4): e00375.
[32] Motegi S, Yokoyama Y, Uchiyama A, Ogino S, Takeuchi Y, Yamada K, Hattori T, Hashizume H, Ishikawa Y, Goto M, Ishikawa O., First Japanese case of atypical progeroid syndrome/atypical Werner syndrome with heterozygousmutation, 2014, 41(12): 1047–1052.
[33] Renard D, Fourcade G, Milhaud D, Bessis D, Esteves-Vieira V, Boyer A, Roll P, Bourgeois P, Levy N, De Sandre-Giovannoli A. Novelmutation in atypical Werner syndrome presenting with ischemic disease, 2009, 40(2): e11–e14.
[34] Mory PB, Crispim F, Freire MB, Salles JE, Valério CM, Godoy-Matos AF, Dib SA, Moisés RS. Phenotypic diversity in patients with lipodystrophy associated withmutations, 2012, 167(3): 423– 431.
[35] Jeru I, Vatier C, Vantyghem MC, Lascols O, Vigouroux C.-associated partial lipodystrophy: anticipation of metabolic complications, 2017, 54(6): 413– 416.
[36] van Tintelen JP, Hofstra RM, Katerberg H, Rossenbacker T, Wiesfeld ACP, du Marchie Sarvaas GJ, Wilde AAM, van Langen IM, Nannenberg EA, van der Kooi AJ, Kraak M, van Gelder IC, van Veldhuisen DJ, Vos Y, van den Berg MP, Working Group on Inherited Cardiac Disorders, line 27/50, Interuniversity Cardiology Institute of The Netherlands. High yield ofmutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics, 2007. 154(6): 1130–1139.
[37] Fountas A, Giotaki Z, Dounousi E, Liapis G, Bargiota A, Tsatsoulis A, Tigas S. Familial partial lipodystrophy and proteinuric renal disease due to a missense c.1045C > Tmutation, 2017, 2017: 0017–0049.
[38] Andre P, Schneebeli S, Vigouroux C, Lascols O, Schaaf M, Chevalier P. Metabolic and cardiac phenotype characterization in 37 atypical Dunnigan patients with nonfarnesylated mutated prelamin A, 2015, 169(4): 587–593.
[39] Muschke P, Kölsch U, Jakubiczka S, Wieland I, Brune T, Wieacker P. The heterozygousmutation p.R471G causes a variable phenotype with features of two types of familial partial lipodystrophy, 2007, 143A(23): 2810–2814.
[40] Saha B, Lessel D, Hisama FM, Leistritz DF, Friedrich K, Martin GM, Kubisch C, Oshima J. A novelmutation causes altered nuclear morphology and symptoms of familial partial lipodystrophy (Dunnigan variety) with progeroid features, 2010, 1(3): 127–132.
[41] Wehnert MS, Feuer A, Wasner C, Hoeltzenbein M. Novel P485R mutation inpresenting with features of a progeroid syndrome., 2004, 14: 591–592.
[42] de Andrade NXS, Adiyaman SC, Yuksel BD, Ferrari CT, Eldin AJ, Saydam BO, Altay C, Sharma P, Bhave N, Little A, McKeever P, Onay H, Ozkal S, Secil M, Yenerel MN, Akinci B, Oral EA. Unusual presentations of- associated lipodystrophy with complex phenotypes and generalized fat loss: when the genetic diagnosis uncovers novel features., 2020, 6(2): e79–e85.
[43] Morel CF, Thomas MA, Cao H, O'Neil CH, Pickering JG, Foulkes WD, Hegele RA. Asplicing mutation in two sisters with severe Dunnigan-type familial partial lipodystrophy type 2, 2006, 91(7): 2689–2695.
[44] Chirico V, FerraùV, Loddo I, Briuglia S, Amorini M, Salpietro V, Lacquaniti A, Salpietro C, Arrigo T.gene mutation as a model of cardiometabolic dysfunction: from genetic analysis to treatment response, 2014, 40(3): 224–228.
[45] Savage DB, Soos MA, Powlson A, O'Rahilly S, McFarlane I, Halsall DJ, Barroso I, Thomas EL, Bell JD, Scobie I, Belchetz PE, Kelly WF, Schafer AJ. Familial partial lipodystrophy associated with compound heterozygosity for novel mutations in thegene, 2004, 47(4): 753–756.
[46] Chan D, McIntyre AD, Hegele RA, Don-Wauchope AC. Familial partial lipodystrophy presenting as metabolic syndrome, 2016, 10(6): 1488–1491.
[47] Guillín-Amarelle C, Sánchez-Iglesias S, Mera A, Pintos E, Castro-Pais A, Rodríguez-Cañete L, Pardo J, Casanueva FF, Araújo-Vilar D. Inflammatory myopathy in the context of an unusual overlapping laminopathy, 2018, 62(3): 376–382.
[48] Patni N, Hatab S, Xing C, Zhou Z, Quittner C, Garg A. A novel autosomal recessive lipodystrophy syndrome due to homozygousvariant, 2020, 57(6): 422–426.
[49] Patni N, Xing C, Agarwal AK, Garg A. Juvenile-onset generalized lipodystrophy due to a novel heterozygous missensemutation affecting lamin C, 2017, 173(9): 2517–2521.
[50] Montenegro Jr RM, Costa-Riquetto AD, Fernandes VO, Montenegro A, de Santana LS, Jorge AAL, Karbage L, Aguiar LB, Carvalho FHC, Teles MG, d'Alva CB. Homozygous and heterozygous nuclear lamin A p.R582C mutation: different lipodystrophic phenotypes in the same kindred, 2018, 9: 458.
[51] Kao KT, Zacharin M. An adolescent girl referred with Cushing syndrome—does she or does she not have the syndrome?, 2016, 29(1): 109– 112.
[52] Garg A, Vinaitheerthan M, Weatherall PT, Bowcock AM. Phenotypic heterogeneity in patients with familial partial lipodystrophy (dunnigan variety) related to the site of missense mutations in lamin a/c gene, 2001, 86(1): 59–65.
[53] Soyaltin UE, Simsir IY, Akinci B, Altay C, Adiyaman SC, Lee K, Onay H, Oral EA. Homozygousp.R582H pathogenic variant reveals increasing effect on the severity of fat loss in lipodystrophy, 2020, 6: 13.
[54] Vigouroux C, MagréJ, Vantyghem MC, Bourut C, Lascols O, Shackleton S, Lloyd DJ, Guerci B, Padova G, Valensi P, Grimaldi A, Piquemal R, Touraine P, Trembath RC, Capeau J. Lamin A/C gene: sex-determined expression of mutations in Dunnigan-type familial partial lipodystrophy and absence of coding mutations in congenital and acquired generalized lipoatrophy, 2000, 49(11): 1958–1962.
[55] Hegele RA, Cao H, Anderson CM, Hramiak IM. Heterogeneity of nuclear lamin A mutations in Dunnigan- type familial partial lipodystrophy, 2000. 85(9): 3431–3435.
[56] Araújo-Vilar D, Lado-Abeal J, Palos-Paz F, Lattanzi G, Bandín MA, Bellido D, Domínguez-Gerpe L, Calvo C, Pérez O, Ramazanova A, Martínez-Sánchez N, Victoria B, Costa-Freitas AT. A novel phenotypic expression associated with a new mutation ingene, characterized by partial lipodystrophy, insulin resistance, aortic stenosis and hypertrophic cardiomyopathy, 2008, 69(1): 61–68.
[57] Resende ATP, Martins CS, Bueno AC, Moreira AC, Foss-Freitas MC, de Castro M. Phenotypic diversity and glucocorticoid sensitivity in patients with familial partial lipodystrophy type 2, 2019, 91(1): 94–103.
[58] Rankin J, Auer-Grumbach M, Bagg W, Colclough K, Nguyen TD, Fenton-May J, Hattersley A, Hudson J, Jardine P, Josifova D, Longman C, McWilliam R, Owen K, Walker M, Wehnert M, Ellard S. Extreme phenotypic diversity and nonpenetrance in families with thegene mutation R644C, 2008, 146A(12): 1530–1542.
[59] Monteiro LZ, Foss-Freitas MC, Júnior Montenegro RM, Foss MC. Body fat distribution in women with familial partial lipodystrophy caused by mutation in the lamin A/C gene, 2012, 16(1): 136–138.
[60] Le Dour C, Schneebeli S, Bakiri F, Darcel F, Jacquemont ML, Maubert MA, Auclair M, Jeziorowska D, Reznik Y, Béréziat V, Capeau J, Lascols O, Vigouroux C. A homozygous mutation of prelamin-A preventing its farnesylation and maturation leads to a severe lipodystrophic phenotype: new insights into the pathogenicity of nonfarnesylated prelamin-A, 2011, 96(5): E856–E862.
[61] Hussain I, Patni N, Garg A. Lipodystrophies, dyslipidaemias and atherosclerotic cardiovascular disease, 2019, 51(2): 202–212.
[62] Lazarte J, Jurgens SJ, Choi SH, Khurshid S, Morrill VN, Weng L-C, Nauffal V, Pirruccello JP, Halford JL, Hegele RA, Ellinor PT, Lunetta KL, Lubitz SA.Variants and Risk of Adult-Onset Cardiac Disease, 2022, 80(1): 50–59.
[63] Glöcklhofer CR, Steinfurt J, Franke G, Hoppmann A, Glantschnig T, Perez-Feliz S, Alter S, Fischer J, Brunner M, Rainer PP, Köttgen A, Bode C, Odening KE. A novelnonsense mutation causes two distinct phenotypes of cardiomyopathy with high risk of sudden cardiac death in a large five-generation family, 2018, 20(12): 2003–2013.
[64] Forissier JF, Bonne G, Bouchier C, Duboscq-Bidot L, Richard P, Wisnewski C, Briault S, Moraine C, Dubourg O, Schwartz K, Komajda M. Apical left ventricular aneurysm without atrio-ventricular block due to a lamin A/C gene mutation, 2003, 5(6): 821–825.
[65] Kumar S, Baldinger SH, Gandjbakhch E, Maury P, Sellal JM, Androulakis AF, Waintraub X, Charron P, Rollin A, Richard P, Stevenson WG, Macintyre CJ, Ho CY, Thompson T, Vohra JK, Kalman JM, Zeppenfeld K, Sacher F, Tedrow UB, Lakdawala NK. Long-term arrhythmic and nonarrhythmic Outcomes of Lamin A/C Mutation carriers, 2016, 68(21): 2299–2307.
[66] van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbüchel H, de Visser M, Crijns HJ, Pinto YM. Meta-analysis of clinical characteristics of 299 carriers ofgene mutations: do lamin A/C mutations portend a high risk of sudden death?, 2005, 83(1): 79–83.
[67] Sidhu K, Castrini AI, Parikh V, Reza N, Owens A, Tremblay-Gravel M, Wheeler MT, Mestroni L, Taylor M, Graw S, Gigli M, Merlo M, Paldino A, Sinagra G, Judge DP, Ramos H, Mesubi O, Brown E, Turnbull S, Kumar S, Roy D, Tedrow UB, Ngo L, Haugaa K, Lakdawala NK., The response to cardiac resynchronization therapy incardiomyopathy, 2022, 24(4): 685–693.
[68] Menezes MP, Waddell LB, Evesson FJ, Cooper S, Webster R, Jones K, Mowat D, Kiernan MC, Johnston HM, Corbett A, Harbord M, North KN, Clarke NF. Importance and challenge of making an early diagnosis in- related muscular dystrophy, 2012, 78(16): 1258–1263.
[69] Spuler S, Kalbhenn T, Zabojszcza J, van Landeghem FK, Ludtke A, Wenzel K, Koehnlein M, Schuelke M, Lüdemann L, Schmidt HH. Muscle and nerve pathology in Dunnigan familial partial lipodystrophy, 2007, 68(9): 677–683.
[70] McPherson E, Turner L, Zador I, Reynolds K, Macgregor D, Giampietro PF. Ovarian failure and dilated cardiomyopathy due to a novel lamin mutation, 2009, 149A(4): 567–572.
[71] Gambineri A, Zanotti L. Polycystic ovary syndrome in familial partial lipodystrophy type 2 (FPLD2): basic and clinical aspects, 2018, 9(1): 392–397.
[72] Thong KM, Xu YX, Cook J, Takou A, Wagner B, Kawar B, Ong AC. Cosegregation of focal segmental glomerulosclerosis in a family with familial partial lipodystrophy due to a mutation in, 2013, 124(1–2): 31–37.
[73] Owen KR, Donohoe M, Ellard S, Clarke TJ, Nicholls AJ, Hattersley AT, Bingham C. Mesangiocapillary glomerulonephritis type 2 associated with familial partial lipodystrophy (Dunnigan-Kobberling syndrome), 2004, 96(2): c35–c38.
[74] Nicolas HA, Akimenko MA, Tesson F. Cellular and animal models of striated muscle laminopathies, 2019, 8(4): 291.
[75] Young SG, Fong LG, Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria—new evidence suggesting that protein farnesylation could be important for disease pathogenesis, 2005, 46(12): 2531– 2558.
[76] Rusiñol AE, Sinensky MS. Farnesylated lamins, progeroid syndromes and farnesyl transferase inhibitors, 2006, 119(Pt 16): 3265–3272.
[77] Broers JLV, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing, 2006, 86(3): 967–1008.
[78] Cau P, Navarro C, Harhouri K, Roll P, Sigaudy S, Kaspi E, Perrin S, De Sandre-Giovannoli A, Lévy N. Nuclear matrix, nuclear envelope and premature aging syndromes in a translational research perspective, 2014, 29: 125–147.
[79] Caron M, Auclair M, Donadille B, Béréziat V, Guerci B, Laville M, Narbonne H, Bodemer C, Lascols O, Capeau J, Vigouroux C. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence, 2007, 14(10): 1759–1767.
[80] Bidault G, Garcia M, Vantyghem MC, Ducluzeau PH, Morichon R, Thiyagarajah K, Moritz S, Capeau J, Vigouroux C, Béréziat V. Lipodystrophy-linkedp.R482W mutation induces clinical early atherosclerosis and in vitro endothelial dysfunction, 2013, 33(9): 2162–2171.
[81] Lloyd DJ, Trembath RC, Shackleton S. A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies, 2002, 11(7): 769–777.
[82] Meyer SU, Sass S, Mueller NS, Krebs S, Bauersachs S, Kaiser S, Blum H, Thirion C, Krause S, Theis FJ, Pfaffl MW. Integrative analysis of microRNA and mRNA data reveals an orchestrated function of microRNAs in skeletal myocyte differentiation in response to TNF-α or IGF1, 2015, 10(8): e0135284.
[83] Tomé M, López-Romero P, Albo C, Sepúlveda JC, Fernández-Gutiérrez B, Dopazo A, Bernad A, González MA. miR-335 orchestrates cell proliferation, migration and differentiation in human mesenchymal stem cells, 2011, 18(6): 985–995.
[84] Oldenburg A, Briand N, Sørensen AL, Cahyani I, Shah A, Moskaug J, Collas P. A lipodystrophy-causing lamin A mutant alters conformation and epigenetic regulation of the anti-adipogenic MIR335 locus, 2017, 216(9): 2731–2743.
[85] Choi JC, Muchir A, Wu W, Iwata S, Homma S, Morrow JP, Worman HJ. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation, 2012, 4(144): 144ra102.
[86] Ramos FJ, Chen SC, Garelick MG, Dai DF, Liao CY, Schreiber KH, MacKay VL, An EH, Strong R, Ladiges WC, Rabinovitch PS, Kaeberlein M, Kennedy BK. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival, 2012, 4(144): 144ra103.
[87] Chartoumpekis DV, Palliyaguru DL, Wakabayashi N, Khoo NK, Schoiswohl G, O'Doherty RM, Kensler TW. Notch intracellular domain overexpression in adipocytes confers lipodystrophy in mice, 2015, 4(7): 543–550.
[88] Perepelina K, Dmitrieva R, Ignatieva E, Borodkina A, Kostareva A, Malashicheva A. Lamin A/C mutation associated with lipodystrophy influences adipogenic differentiation of stem cells through interaction with Notch signaling, 2018, 96(3): 342– 348.
[89] Le Dour C, Wu W, Béréziat V, Capeau J, Vigouroux C, Worman HJ. Extracellular matrix remodeling and transforming growth factor-β signaling abnormalities induced by lamin A/C variants that cause lipodystrophy, 2017, 58(1): 151–163.
[90] Muchir A, Medioni J, Laluc M, Massart C, Arimura T, van der Kooi AJ, Desguerre I, Mayer M, Ferrer X, Briault S, Hirano M, Worman HJ, Mallet A, Wehnert M, Schwartz K, Bonne G. Nuclear envelope alterations in fibroblasts from patients with muscular dystrophy, cardiomyopathy, and partial lipodystrophy carrying lamin A/C gene mutations, 2004, 30(4): 444–450.
[91] Vigouroux C, Guénantin AC, Vatier C, Capel E, Le Dour C, Afonso P, Bidault G, Béréziat V, Lascols O, Capeau J, Briand N, Jéru I. Lipodystrophic syndromes due tomutations: recent developments on biomolecular aspects, pathophysiological hypotheses and therapeutic perspectives, 2018, 9(1): 235–248.
[92] Foss-Freitas MC, Akinci B, Luo YY, Stratton A, Oral EA. Diagnostic strategies and clinical management of lipodystrophy, 2020, 15(2): 95–114.
[93] Vatier C, Fetita S, Boudou P, Tchankou C, Deville L, Riveline J, Young J, Mathivon L, Travert F, Morin D, Cahen J, Lascols O, Andreelli F, Reznik Y, Mongeois E, Madelaine I, Vantyghem M, Gautier J, Vigouroux C. One-year metreleptin improves insulin secretion in patients with diabetes linked to genetic lipodystrophic syndromes, 2016, 18(7): 693–697.
[94] Grundfest-Broniatowski S, Yan JL, Kroh M, Kilim H, Stephenson A. Successful treatment of an unusual case of FPLD2: the role of Roux-en-Y gastric bypass-case report and literature review, 2017, 21(4): 739–743.
[95] Boettcher E, Csako G, Pucino F, Wesley R, Loomba R. Meta-analysis: pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis, 2012, 35(1): 66–75.
[96] Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, Van Natta M, Clark J, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR, NASH CRN. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis, 2010, 362(18): 1675–1685.
Advances in lipodystrophy syndrome caused bygene mutation
Cheng Xiao1, Jieying Liu1,2, Chunru Yang1, Miao Yu1
Lipodystrophy syndrome caused bygene mutation is a group of autosomal dominant monogenic diseases, characterized by selective fat loss and metabolic abnormalities with insulin resistance. In this review, we summarize the clinical manifestations caused by multiple pathogenicmutations reported so far, including metabolic complications, cardiovascular abnormalities, gonadal axis disorders, myopathy, and renal abnormalities. Meanwhile, we also clarify the possible pathogenic mechanism, diagnosis, and treatment, in order to improve the understanding of the disease and to provide a reference for basic research and clinical diagnosis and treatment of this disease.
lipodystrophy syndrome;gene mutation; insulin resistance; metabolic disorders; pathogenic mechanisms
2022-06-30;
2022-09-06;
2022-09-20
国家自然科学基金项目(编号:82170855)和科技部国家重点研发计划项目(编号:2020YFC2004505,2018YFC2001105)资助[Supported by the National Nature Science Foundation of China (No. 82170855) and the National Key Research and Development Program (Nos. 2020YFC2004505, 2018YFC2001105)]
肖诚,在读博士研究生,专业方向:内分泌与代谢病方向。E-mail: jz_xiaocheng@163.com
于淼,博士,教授,研究方向:内分泌与代谢病方向。E-mail: yumiaoxh@163.com
10.16288/j.yczz.22-225
(责任编委: 周红文)
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
英语世界(2022年9期)2022-10-18 01:11:24
保健医苑(2022年4期)2022-05-05 06:11:14
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
肝博士(2021年1期)2021-03-29 02:32:10
中国生殖健康(2020年2期)2021-01-18 02:51:26
小哥白尼(趣味科学)(2020年1期)2020-06-16 03:24:44
生物学通报(2019年3期)2019-02-17 18:03:58
小学生导刊(2018年13期)2018-06-29 03:49:00
