
BSCL2基因复合杂合突变导致先天性全身性脂肪萎缩的分子机制研究
2022-11-21 03:19叶静雅黄爱洁付真真龚颖芸杨洪远周红文
遗传 2022年10期
叶静雅,黄爱洁,付真真,龚颖芸,杨洪远,周红文
研究报告
基因复合杂合突变导致先天性全身性脂肪萎缩的分子机制研究
叶静雅1,黄爱洁1,付真真1,龚颖芸1,杨洪远2,周红文1
1. 南京医科大学第一附属医院内分泌科,南京 210029 2. 澳大利亚新南威尔士大学生物技术与生物分子科学学院,悉尼 999029,澳大利亚
先天性全身性脂肪萎缩(congenital generalized lipodystrophy,CGL)是一种极端罕见的常染色体隐性遗传病,表现为明显的全身脂肪极度缺失,肌肉感明显,并伴有一系列的代谢指标异常,包括严重的胰岛素抵抗,高血糖,高脂血症,脂肪肝以及黑棘皮等。本文针对1例CGL患者及其家系进行研究。先证者为19岁年轻女性,自幼皮下脂肪缺如,血清瘦素水平仅0.14 μg/L。对患者及其亲属(父母、弟弟)进行全基因组检测,显示该患者基因5号外显子存在复合杂合突变(c.560A>G和c.565G>T),c.560A>G突变导致对应编码的187位的氨基酸由酪氨酸突变为半胱氨酸(p.Y187C),从而引起编码的SEIPIN蛋白发生错义突变;c.565 G>T突变引起对应编码的189位氨基酸转为终止密码子(p.E189X),产生蛋白截短突变。经Sanger测序验证,患者父亲携带c.565G>T杂合突变,患者母亲携带c.560A>G杂合突变,患者弟弟未携带基因致病性突变。本研究通过转染突变p.Y187C质粒至HEK293细胞,观察到SEIPIN蛋白量及与甘油-3-磷酸酰基转移酶(glycerol-3-phosphate acyltransferase, GPAT3)互作减少;原代培养的患者皮肤成纤维细胞体外功能实验表明,患者的SEIPIN蛋白量明显低于正常健康人,加入组蛋白去乙酰化酶抑制剂(histone deacetylase inhibitors, HDACis)可部分挽救SEIPIN蛋白表达。此外,油酸刺激下患者皮肤成纤维细胞脂滴小于正常健康人。本文同时综述国内外既往文献中报道的基因突变位点,丰富了CGL的临床表型谱和致病基因突变谱,有助于提高临床医生对CGL的临床诊治和致病机制的理解。
先天性全身性脂肪萎缩;/SEIPIN;成纤维细胞;HDACi;糖尿病
先天性全身性脂肪萎缩(congenital generalized lipodystrophy, CGL),又称贝赛综合征(Berardinelli- Seip syndrome),是一种极端罕见的常染色体隐性遗传病,发病率约为1/10,000,000[1]。CGL的患者表现为明显的全身脂肪缺如,肌肉感明显,并伴有一系列的代谢指标异常,包括严重的胰岛素抵抗,高血糖,高脂血症,脂肪肝以及黑棘皮等[2,3]。此外,在CGL患者血中来自脂肪细胞分泌的瘦素和脂联素水平低下,平均约为0.63 ng/mL和1.5 μg/mL[4],正常人血清瘦素和脂联素的平均含量分别为16 ng/mL 和6.8 μg/mL[5,6]。
临床上根据突变基因不同将CGL分为4类,分别为CGL1(1-acylglycerol-3-phosphate O-acyltransferase 2,基因突变),CGL2(Berardinelli-Seip congenital lipodystrophy 2,基因突变),CGL3 (caveolin 1,基因突变)和CGL4(polymerase I and transcript release factor,基因突变)。基因突变导致的2型CGL是所有类型中最严重的,部分患者会伴发心肌病、神经迟滞、早死和智力缺损[7,8]。CGL2型的突变基因位于人13号染色体上,编码一种跨内质网膜保守蛋白SEIPIN。SEIPIN在大脑,睾丸,脂肪组织中高表达[9,10],在脂滴融合、聚集与扩大的过程中起重要作用[3,11~13]。相关研究表明通过调控中枢下丘脑–垂体轴以及外周脂肪细胞分化来影响脂质储存过程,基因突变的哺乳动物细胞显现出异常的内质网–脂滴接触以及增多的小脂滴[14,15],此外,临床报道中,突变的CGL2患者中也表现出严重的脂肪组织萎缩[2]。这些结果提示基因编码的SEIPIN蛋白有着显著调控细胞脂滴融合的能力。
CGL2于20世纪50年代首次报道[16],后陆续在美洲、欧洲、非洲、亚洲等多个种族人群中发现[7,17~19]。迄今为止,中国仅有少数病例报道[20~22]。本文报道1例先天性全身性脂肪萎缩的中国女性患者,通过全基因组测序技术发现其携带基因复合杂合突变(c.560A>G, p.Y187C和c.565G>T, p.E189X),利用患者皮肤成纤维细胞进行体外功能实验,结果表明患者来源的细胞中SEIPIN蛋白减少,脂肪酸刺激下细胞中形成的脂滴形态偏小,加入组蛋白去乙酰化酶抑制剂(histone deacetylase inhibitors, HDACis)可部分挽救SEIPIN蛋白表达,最后,本文还总结了既往文献中报道的基因突变位点。
1 对象与方法
1.1 对象及临床资料收集
患者19岁,女性,因“自幼皮下脂肪缺如伴血糖升高1年”来本院就诊。患者就诊前1年出现多饮多尿症状,于当地县医院诊断“糖尿病”,空腹血糖在15~20 mmol/L之间波动,患者在疾病过程中,未出现酮症酸中毒。初次本院就诊体格检查:身高160 cm,体重46 kg,BMI 17.9 kg/m2,神清,精神可,全身皮下脂肪缺如,可见明显的二头肌、四头肌和小腿肌肉、肌腱和皮下静脉,黑棘皮,多毛,肝脏在右锁骨中线右肋下2 cm处可触及,脾在左锁骨中线左肋下3cm处可见,血压正常,四肢活动正常,心肺检查未见明显异常,神经系统检查无明显异常。父母及其弟弟体重正常,无明显消瘦。总胆固醇、甘油三酯、高密度脂蛋白胆固醇(HDL-c)、低密度脂蛋白胆固醇(LDL-c)、血糖、糖化血红蛋白(HbA1c)、促甲状腺激素全套、口服糖耐量实验(oral glucose tolerance test, OGTT)、高胰岛素正糖钳夹(评估胰岛素敏感性)、血清瘦素均在南京医科大学第一附属医院检验实验室检测。患者及家属均已签署知情同意书,本研究方案经南京医科大学第一附属医院机构审查委员会批准。
1.2 全外显子测序及验证
患者及其父母,弟弟的基因组DNA均提取自采集的外周血液样本,提取的DNA送至北京诺禾致源公司进行基因全外显子测序,测序返回的结果进行Sanger一代测序验证,基因序列与GenBank(Gene ID: 26580)进行比对,测序结果用Sanger测序验证,基因5号外显子扩增测序引物序列,正向引物:5′-AGGGACCTGAGGAAGAAG-3′,反向引物5′-AGAATCCTGGGCTAATGG-3′。
1.3 细胞培养及HDACi挽救实验
患者及年龄性别匹配正常人对照皮肤成纤维细胞提取步骤如前所述[23]。细胞培养在高糖DMEM (Gibco,美国)培养基中,含有10%胎牛血清及1%双抗。HDACi药物细胞实验,SAHA (suberoylanilide hydroxamic acid)、帕比司他(Panobinostat)均购于美国MCE公司,加药浓度为2.5 μmol/L,加药时间48 h。
1.4 蛋白质印记(Western blot)和免疫沉淀(immunoprecipitation, IP)实验
兔抗人源SEIPIN抗体由河南医科大学的高明明教授团队惠赠,羊抗兔二抗购买于Abcam公司(美国),使用稀释度为1∶2000。免疫沉淀实验使用磁珠吸附法(Invitrogen, 美国)。贴壁细胞从细胞培养皿中刮取收集,加入IP裂解液,细胞蛋白4℃高速离心后收集,加入相应一抗及磁珠孵育过夜。用重悬试剂将抗体结合的蛋白重新与磁珠分离,并后续进行Western blot实验。
1.5 免疫荧光(immunofluorescence, IF)实验
成纤维细胞种在玻片上,用4%的多聚甲醛固定15 min后,用PBS缓冲液清洗3遍,再用0.2% Triton X-100固定5 min,继续用含有1%BSA的PBS封闭1 h,分别加入一抗4℃过夜,PBS洗3遍,荧光二抗避光孵育2 h后,添加荧光抗淬剂封片并在荧光显微镜下成像拍片。
1.6 统计分析
数据以均数±标准差方式展现,统计分析方法以Student’s-test和方差分析。
2 结果与分析
2.1 患者的临床表现
除了显著的脂肪萎缩症状(图1A),核磁共振成像(magnetic resonance imaging, MRI)及双能X线(dual energy X-ray absorptiometry, DEXA)结果显示,CGL病人表型出明显的皮下脂肪缺如(图1,B和D),而患者骨密度没有明显改变(图1C)。患者血清甘油三酯,糖化血红蛋白均升高,分别为2.3 mmol/L和10.6%,同时伴有胰岛素抵抗(空腹胰岛素1001 pmol/L),高血糖(空腹血糖16.37 mmol/L),患者血清瘦素极低,仅只有0.14 μg/L血清胆固醇、HDL-c、FT3、FT4没有明显异常(表1)。OGTT证明该患者血糖升高伴随胰岛素抵抗,而患者父母及亲弟弟血清葡萄糖水平与糖刺激下胰岛素分泌正常(图1G)。除此以外,高胰岛素正糖钳夹技术发现患者胰岛素敏感性指标葡萄糖输注率(glucose infusion rate, GIR)仅为1.6 mg/kg/min,低于课题组前期究队列里正常的中国成年人(平均GIR为10.9± 2.25 mg/kg/min)。这些结果均提示患者因全身性脂肪缺失而伴有严重的胰岛素抵抗症状。
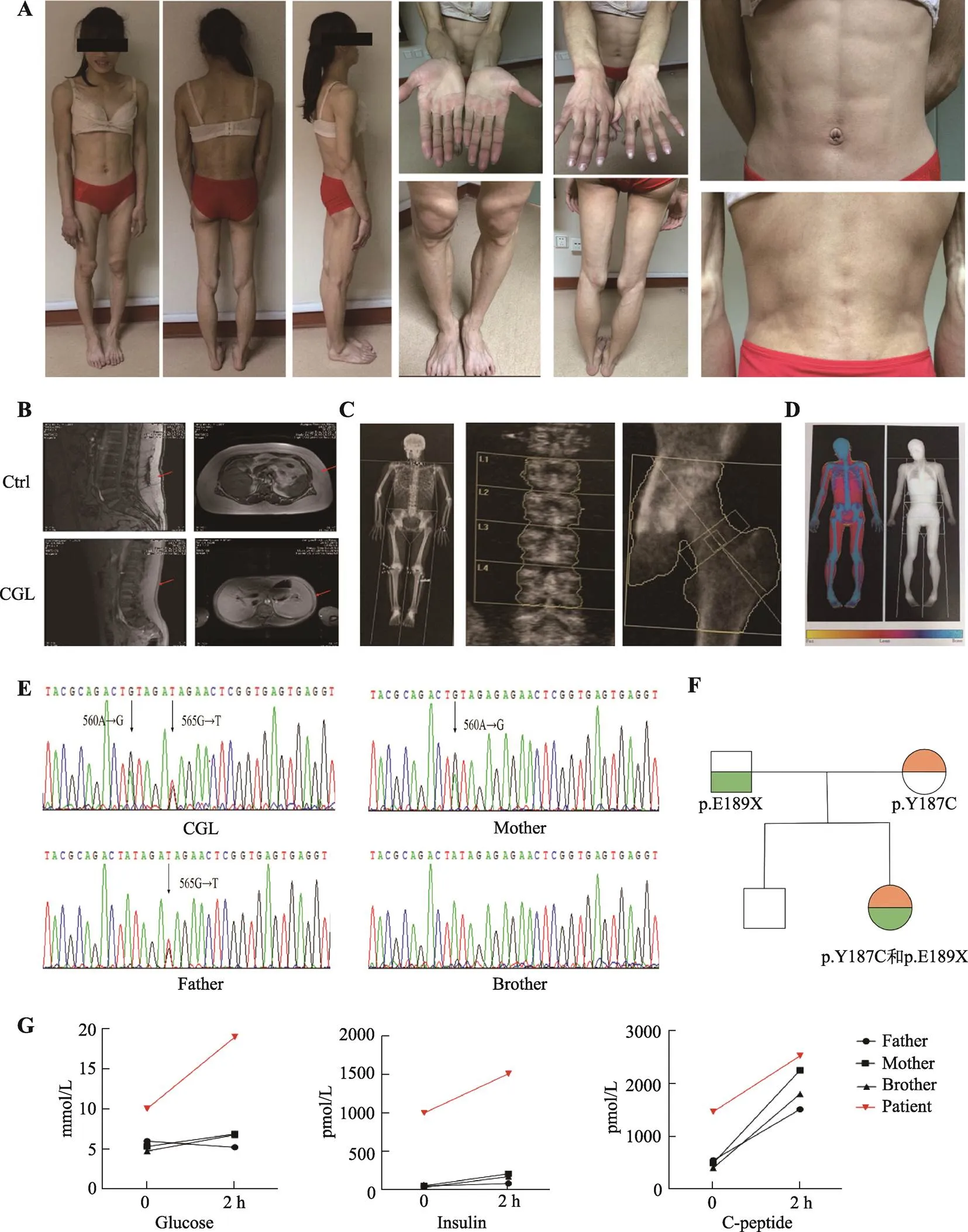
图1 CGL患者临床表型
A:CGL患者从正面、背面和侧面拍摄的照片,以及手臂、腿、手指、脚、腹部和后腰部的照片。B:患者和肥胖对照受试者的腹部MRI图像。红色箭头:腹部脂肪组织。C:骨密度图像,L1~L4腰椎和髋骨。D:患者体脂成分分析。黄色,脂肪;红色:肌肉等瘦组织;蓝色:骨骼。E:c.560A>G和c.565G>C突变的测序结果。F:CGL患者的家族谱系。绿色:p.E189X突变,橙色:p.Y187C突变。正方形:男性,圆形:女性。G:OGTT 观察患者及家属的血糖、胰岛素、C肽。
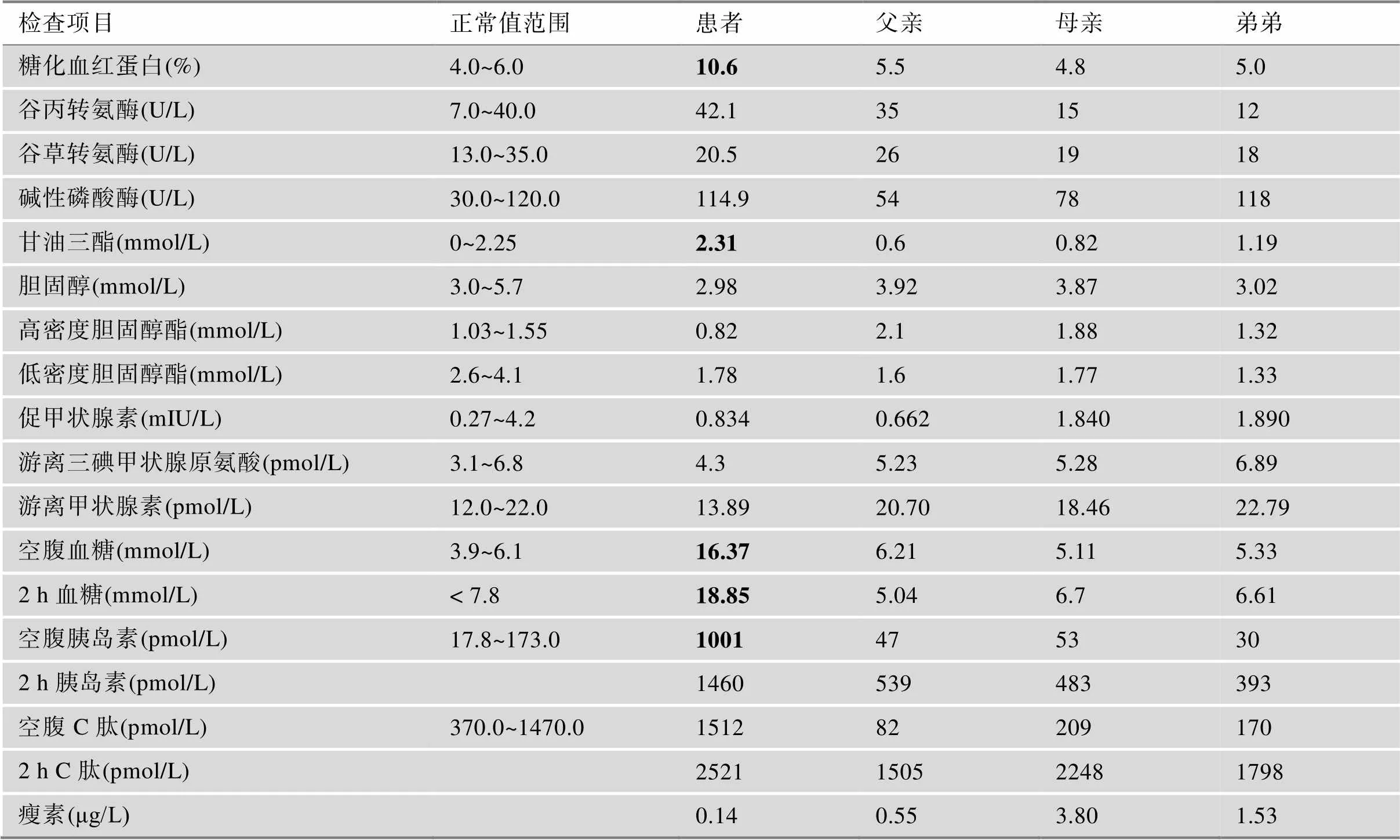
表1 CGL家系实验室检查指标
加粗数值为异常值。
2.2 基因检测结果分析显示先证者携带BSCL2基因复合杂合突变
全外显子检测显示先证者携带基因5号外显子复合杂合突变(c.560A>G, p.Y187C和c.565G>T, p.E189X),患者父亲携带c.565G>T(p.E189X)突变,患者母亲携带c.560A>G(p.Y187C)突变,患者弟弟未携带基因致病性突变,符合常染色体隐性遗传致病模式(图1,E和F)。c.560A>G突变表现为外显子5的560位点的腺嘌呤突变为鸟嘌呤,从而引起错义突变,对应编码的187位的氨基酸有由酪氨酸突变为半胱氨酸(p.Y187C),查阅1000G、ESP、ExAc等数据库,该突变位点位于热点突变区域,与关键酶的活性位点相关,且人群突变频率极低,ACMG评级为可能致病突变。c.565G>T突变表现为565位鸟嘌呤突变为胸腺嘧啶,从而引起截短突变,对应编码的189位氨基酸转为终止密码子而停止翻译,ACMG评级为致病突变。
2.3 蛋白定量与功能实验验证突变细胞脂滴变小
SEIPIN蛋白错义突变p.Y187C位点尽管数年前在日本人群中被报道,然而相关蛋白功能没有得到进一步验证。课题组在HEK293工具细胞中转染携带HA标签的p.Y187C突变质粒。转染p.Y187C质粒的细胞内SEIPIN含量明显减少(图2A),免疫荧光结果进一步提示携带突变质粒的细胞SEIPIN蛋白分布异常(图2B)。澳大利亚杨洪远教授等报道了甘油-3-磷酸酰基转移酶(glycerol-3-phosphate acyltransferase,GPAT3)与SEIPIN蛋白互作共同调控脂滴的大小[24],因此,本研究进一步通过免疫共沉淀观察突变蛋白与GPAT3的互作能力,结果表明突变SEIPIN蛋白导致两者的结合减少(图2C)。此外,来自CGL患者的皮肤成纤维细胞SEIPIN蛋白含量也明显减少(图2,E和G)。为了进一步验证SEIPIN蛋白对脂滴融合能力的影响,用油酸(oleic acid, OA)刺激原代培养的患者及健康人的皮肤成纤维细胞,结果表明患者的成纤维细胞中形成的脂滴变小,融合能力减弱(图2,D和F)。
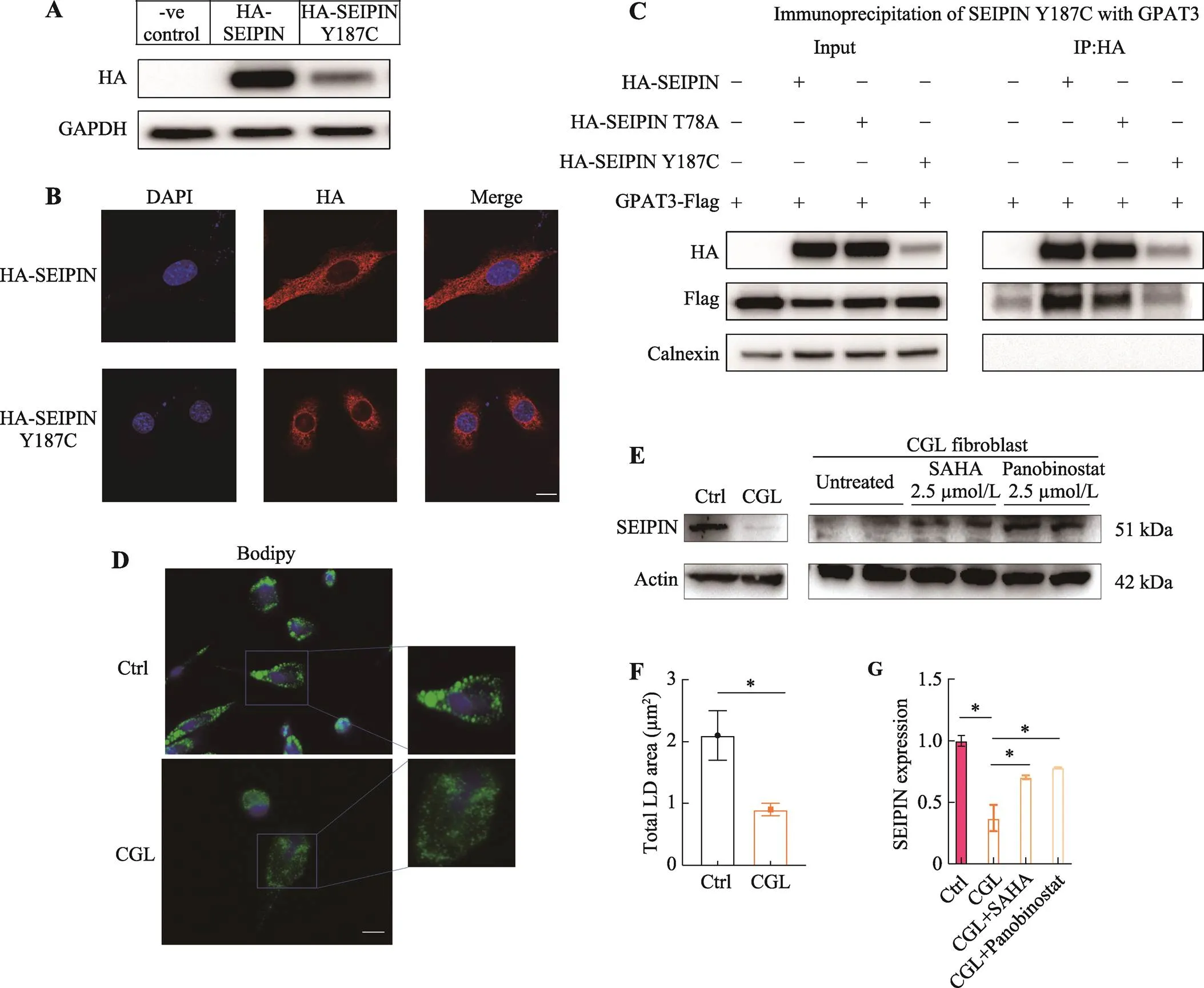
图2 SEIPIN突变的功能特性
A和B:携带HA标签p.Y187C 质粒及野生型质粒转染HEK293细胞后进行蛋白质印迹(A)和免疫荧光观察(B)结果。蓝色:DAPI,红色:HA-SEIPIN。标尺:5 μm。C:免疫沉淀HA标记的SEIPIN的蛋白质印迹及其与标记的GPAT3的相互作用。p.T78A:无义突变;p.Y187C:腺嘌呤到鸟嘌呤替换突变。D:OA刺激后,来自年龄性别匹配的健康志愿者(Ctrl)和CGL患者的皮肤成纤维细胞脂滴染色。蓝色:DAPI;绿色:Bodipy。标尺:5 μm。E:蛋白质印迹观察HDACis (SAHA或Panobinostat)处理48 h的人成纤维细胞中SEIPIN含量。F和G:脂滴染色(D)和蛋白印迹(E)统计分析图,*<0.05。
2.4 组蛋白去乙酰化酶抑制剂挽救实验证明HDACi可部分挽救SEIPIN蛋白表达
HDACi可以通过调控热休克反应与未折叠蛋白的敏感性,减少泛素通路以及溶酶体通路对于错误折叠蛋白的降解,HDACi在其他罕见及常见代谢病中挽救部分被错误折叠的蛋白有效性得到证实,如尼曼–皮克病(Niemann-Pick Disease)[25,26],囊性纤维化[27],以及2型糖尿病[28]。目前文献中并没有报道过HDACi用于治疗CGL,本研究使用HDACi(SAHA及帕比司他)处理患者来源的成纤维细胞,并观察SEIPIN蛋白的稳定性,结果表明HDACi可部分挽救SEIPIN蛋白表达(图2,E和G)。
2.5 BSCL2基因不同位点突变引起不同疾病
基于本病例的发现,本研究总结了1954~2021年的CGL2型病例基因突变位点,并绘制了基因-SEIPIN蛋白关联图(图3)。SEIPIN蛋白由398个氨基酸组成,包括胞质N末端(1~26氨基酸)、C末端(264~398氨基酸)、两个跨膜区(26~48和242~264氨基酸)和一个内质网保守中心环(48~242氨基酸)。总共约有140个不同的突变位点,其中65个突变位于SEIPIN蛋白内质网膜腔面侧,58个突变位于蛋白内质网胞浆侧以及17个突变位于蛋白跨膜区。涉及的病种包括:先天性全身性脂肪萎缩;远端遗传性运动神经元病(distal hereditary motor neuropathies,DHMN);腓骨肌萎缩症(Charcot- Marie-Tooth disease, DMT)以及多种疾病并存。同时,本研究统计了不同大洲的突变频率,美洲最多(70%),其次为欧洲(20%),亚洲(9%),大洋洲(1%)。
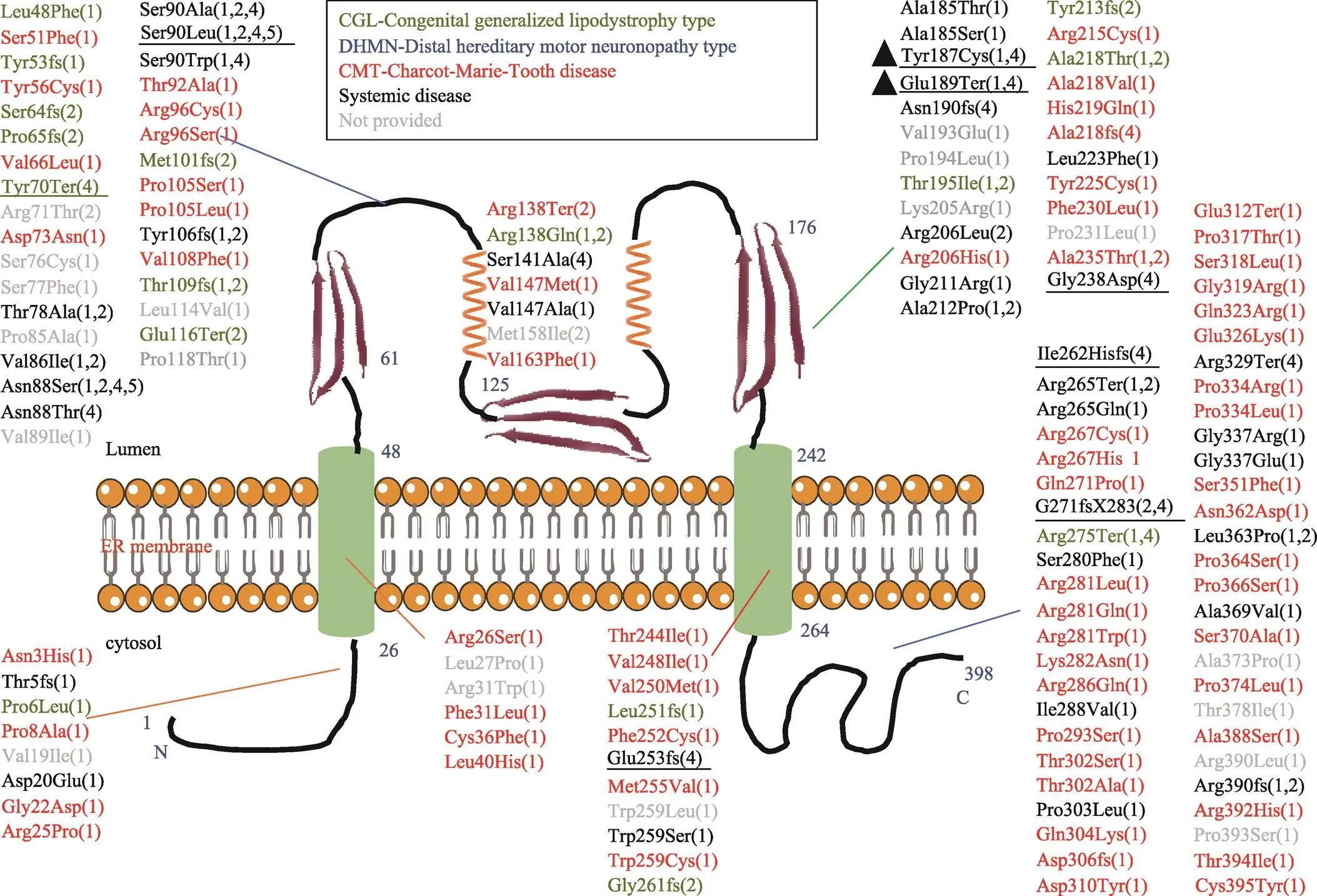
图3 BSCL2基因突变位点到SEIPIN蛋白及临床疾病的关联图
SEIPIN蛋白包括胞质N末端(1~26氨基酸)、C末端(264~398氨基酸)、两个跨膜区(26~48和242~264氨基酸)和一个内质网保守中心环(48~242氨基酸)。颜色表示疾病的类别,绿色:先天性全身性脂肪萎缩;蓝色:远端遗传性运动神经元病;红色:腓骨肌萎缩症;黑色:多种疾病并存;灰色:未找到相应的临床疾病。数字代表不同的大陆。(1):美洲;(2):欧洲;(3):非洲;(4):亚洲;(5):大洋洲。下划线:中国报道的病例。三角:本例患者突变位点。
3 讨论
本文报道了1例先天性全身性脂肪萎缩年轻女性病例,患者因“自幼皮下脂肪缺如伴血糖升高”就诊,血清瘦素水平极低仅有0.14 μg/L。全身皮下脂肪缺如,可见明显的二头肌、四头肌和小腿肌肉、肌腱和皮下静脉,同时伴有胰岛素抵抗,高血糖,高脂血症,黑棘皮,多毛。携带基因复合杂合突变(c.560A>G, p.Y187C和c.565G>T, p.E189X),体外实验证明,突变导致SEIPIN含量减少,降解增多,HDACi可以增加突变蛋白稳定性,保留部分突变蛋白功能。
中国人口庞大,CGL病例报道却仅有数例,提示罕见基因突变引起的CGL疾病存在漏诊或误诊的现象,如何区分单纯营养不良、慢性消耗性疾病如糖尿病,肿瘤造成的脂肪萎缩,对于有效诊治CGL至关重要。临床上存在4中类型的CGL,由基因突变导致的CGL2是最常见的一种[29]。CGL2几乎丢失了全身性的脂肪组织。这些患者被报道部分患有心肌病、神经迟滞、早死和智力缺损、较低的血瘦素和脂联素水平[7,30]。Nishiyama等[31]在2009年报道了一名3个月的患有CGL的日本婴儿携带p.Y187C纯合突变。2007中国最早报道了一个7岁CGL男孩携带SEIPIN蛋白p.E189X纯合突变,该男孩表现出皮下脂肪普遍缺乏、明显的肌肉征、脸颊凹陷、黑棘皮、背部多毛、肝脾肿大、阴茎硬结、心肌功能异常、左心室肥大、轻度智力低下,实验室检查显示患儿有胰岛素抵抗、高甘油三酯血症和脂肪肝。这些症状与本文的CGL患者十分相似,然而也存在部分不同,本文患者的智力和神经系统方面也未见明显异常。由此推测,相较于p.E189X截断突变,p.Y187C错义突变可能仍然保留一些 SEIPIN 功能。
SEIPIN蛋白具有398个氨基酸,包含胞质N端、C端、两个跨膜区和在内质网(ER)腔中的一个保守的中心环。基因(c.565G>T)的突变导致 189 位的翻译终止密码子而不是正常的谷氨酸 (E189X)。因此,推测无义突变 E189X导致截短的蛋白质丢失后续209个氨基酸,引起ER腔面、第二跨膜结构域区和C端结构区缺失,蛋白功能完全丧失。错义突变p.Y187C为SEIPIN蛋白中的单个氨基酸变化,p.Y187C 位于保守的中央环,类似于A212P结构。当在细胞中表达时,与在相同启动子下表达的野生型SEIPIN蛋白相比,Y187C的蛋白水平显着降低,这些结果表明p.Y187C突变体可能会影响SEIPIN蛋白的稳定性。
脂肪营养不良的治疗包括生活方式干预和个体化治疗,目前CGL尚无特效疗法,主要以对症治疗为主。贝特类降脂药适用于患有极度高甘油三酯血症的患者[32],应监测本研究患者的甘油三酯水平以确定贝特类降脂药的必要性。合并糖尿病的CGL患者必须进行降糖治疗。大多数CGL患者可能需要大剂量的胰岛素,以及包括二甲双胍和磺脲类药物在内的口服药物[33]。此外,还需要考虑糖尿病的并发症。住院期间,患者接受了胰岛素泵治疗,最大剂量为每天126 IU。空腹血糖5~6 mmol/L,餐后血糖5~9 mmol/L。出院后,目前接受门冬胰岛素(18U-18U-16U)和甘精胰岛素(72U)治疗。空腹血糖在5~6 mmol/L之间波动,餐后2 h血糖在7~ 8 mmol/L之间波动。瘦素由脂肪组织分泌,CGL2患者的血清瘦素水平明显低于健康个体[30],基于美曲普汀(人类瘦素的重组类似物)的瘦素疗法备受关注。美曲普汀已被证明可以显著改善代谢紊乱,包括糖尿病、高甘油三酯血症和肝脂肪变性[33],以及显著改善脂肪营养不良和严重瘦素缺乏症患者的糖脂代谢[34~36],然而,这些治疗仅降低血浆和肝脏甘油三酯水平,针对血糖改善仍较局限[37]。
除了对症治疗,寻找新的药物来减少突变蛋白的降解也至关重要。组蛋白脱乙酰酶通过组蛋白、转录因子和伴侣蛋白(包括Hsp90)的翻译后修饰来调节细胞功能[38]。通过改变这些蛋白质的乙酰化,HDACis可以调节蛋白质在热休克反应中的基因表达,并减少泛素化和蛋白酶体降解以恢复错误折叠蛋白质的功能。SAHA是第一个获批用于临床治疗晚期皮肤T细胞淋巴瘤的HDACi[39]。据报道,HDACis可通过维持部分突变的蛋白质活性来治疗包括戈谢病和尼曼–皮克病在内的罕见疾病[40,41]。本文还研究了SAHA和帕比司他这两种临床获批的HDACis,体外使用的药物浓度与患者的SAHA血清浓度一致[42]。用SAHA或Panobinostat处理的患者来源的成纤维细胞的蛋白质印迹分析显示SEIPIN蛋白显著增加。然而,这种效应尚未在人体中得到证实,结合现有报道,HDACis治疗CGL的有效性仍需进一步探讨。
综上所述,本文报道了1例携带基因复合杂合突变(p.Y187C和p.E189X)的CGL2患者。患者表现出严重的脂肪营养不良与高血糖,但没有明显的心肌病及智力低下。基因突变导致SEIPIN蛋白含量下降、分布异常,HDACis可部分挽救SEIPIN蛋白表达。本研究丰富了CGL2的临床表型谱,并总结了CGL2的致病基因突变谱,有助于提高临床医生对CGL2的诊治水平和对致病机制的理解。
[1] Agarwal AK, Garg A. Genetic disorders of adipose tissue development, differentiation, and death., 2006, 7: 175–199.
[2] Agarwal AK, Garg A. Genetic basis of lipodystrophies and management of metabolic complications., 2006, 57: 297–311.
[3] Fei WH, Du XM, Yang HY. Seipin, adipogenesis and lipid droplets., 2011, 22(6): 204–210.
[4] Haque WA, Shimomura I, Matsuzawa Y, Garg A. Serum adiponectin and leptin levels in patients with lipodystrophies., 2002, 87(5): 2395.
[5] Farooqi IS, Keogh JM, Kamath S, Jones S, Gibson WT, Trussell R, Jebb SA, Lip GY, O’Rahilly S. Partial leptin deficiency and human adiposity., 2001, 414(6859): 34–35.
[6] Pareja-Galeano H, Santos-Lozano A, Sanchis-Gomar F, Fiuza-Luces C, Garatachea N, Gálvez BG, Lucia A, Emanuele E. Circulating leptin and adiponectin concentrations in healthy exceptional longevity., 2017, 162: 129–132.
[7] Van Maldergem L, Magré J, Khallouf TE, Gedde-Dahl T, Delépine M, Trygstad O, Seemanova E, Stephenson T, Albott CS, Bonnici F, Panz VR, Medina JL, Bogalho P, Huet F, Savasta S, Verloes A, Robert JJ, Loret H, De Kerdanet M, Tubiana-Rufi N, Mégarbané A, Maassen J, Polak M, Lacombe D, Kahn CR, Silveira EL, D'Abronzo FH, Grigorescu F, Lathrop M, Capeau J, O'Rahilly S. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy., 2002, 39(10): 722–733.
[8] Agarwal AK, Simha V, Oral EA, Moran SA, Gorden P, O'Rahilly S, Zaidi Z, Gurakan F, Arslanian SA, Klar A, Ricker A, White NH, Bindl L, Herbst K, Kennel K, Patel SB, Al-Gazali L, Garg A. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy., 2003, 88(10): 4840–4847.
[9] Agarwal AK, Garg A. Seipin: a mysterious protein., 2004, 10(9): 440–444.
[10] Jiang M, Gao MM, Wu CM, He H, Guo XJ, Zhou ZM, Yang HY, Xiao XH, Liu G, Sha JH. Lack of testicular seipin causes teratozoospermia syndrome in men., 2014, 111(19): 7054–7059.
[11] Magré J, Delépine M, Khallouf E, Gedde-Dahl T, Van Maldergem L, Sobel E, Papp J, Meier M, Mégarbané A, Bachy A, Verloes A, d'Abronzo FH, Seemanova E, Assan R, Baudic N, Bourut C, Czernichow P, Huet F, Grigorescu F, de Kerdanet M, Lacombe D, Labrune P, Lanza M, Loret H, Matsuda F, Navarro J, Nivelon-Chevalier A, Polak M, Robert JJ, Tric P, Tubiana-Rufi N, Vigouroux C, Weissenbach J, Savasta S, Maassen JA, Trygstad O, Bogalho P, Freitas P, Medina JL, Bonnicci F, Joffe BI, Loyson G, Panz VR, Raal FJ, O'Rahilly S, Stephenson T, Kahn CR, Lathrop M, Capeau J, BSCL Working Group. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13., 2001, 28(4): 365–370.
[12] Szymanski KM, Binns D, Bartz R, Grishin NV, Li WP, Agarwal AK, Garg A, Anderson RGW, Goodman JM. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology., 2007, 104(52): 20890–20895.
[13] Fei WH, Shui GH, Gaeta B, Du XM, Kuerschner L, Li P, Brown AJ, Wenk MR, Parton RG, Yang HY. Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast., 2008, 180(3): 473–482.
[14] Salo VT, Belevich I, Li SQ, Karhinen L, Vihinen H, Vigouroux C, Magré J, Thiele C, Hölttä-Vuori M, Jokitalo E, Ikonen E. Seipin regulates ER-lipid droplet contacts and cargo delivery., 2016, 35(24): 2699–2716.
[15] Wang HJ, Becuwe M, Housden BE, Chitraju C, Porras AJ, Graham MM, Liu XN, Thiam AR, Savage DB, Agarwal AK, Garg A, Olarte MJ, Lin QQ, Fröhlich F, Hannibal- Bach HK, Upadhyayula S, Perrimon N, Kirchhausen T, Ejsing CS, Walther TC, Farese RV. Seipin is required for converting nascent to mature lipid droplets., 2016, 5: e16582.
[16] BERARDINELLI W. An undiagnosed endocrinometabolic syndrome: report of 2 cases., 1954, 14(2): 193–204.
[17] Magré J, Delépine M, Van Maldergem L, Robert JJ, Maassen JA, Meier M, Panz VR, Kim CA, Tubiana-Rufi N, Czernichow P, Seemanova E, Buchanan CR, Lacombe D, Vigouroux C, Lascols O, Kahn CR, Capeau J, Lathrop M. Prevalence of mutations in AGPAT2 among human lipodystrophies., 2003, 52(6): 1573–1578.
[18] Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation., 2011, 12(11): 722–734.
[19] Miranda DM, Wajchenberg BL, Calsolari MR, Aguiar MJ, Silva JMCL, Ribeiro MG, Fonseca C, Amaral D, Boson WL, Resende BA, De Marco L. Novel mutations of theand AGPAT2 genes in 10 families with Berardinelli-Seip congenital generalized lipodystrophy syndrome., 2009, 71(4): 512–517.
[20] Qin YY, Zhang X, Xiang LQ, Shan QW, Li SD, Yan J, Lin FQ. A new compound heterozygous mutation ofin a chinese zhuang ethnic family with congenital generalized lipodystrophy., 2019, 12: 2583–2587.
[21] Yang Y, Ma L, Sun JJ, Gong XH, Cai C, Hong WC. The neonatal onset diabetes mellitus of Chinese neonate with congenital generalized lipodystrophy 2: a case report., 2022, 22(1): 83.
[22] Wang MF, Cun ZK, Peng JC, Chen R, Li JW. Type 2 congenital generalized lipodystrophy with a heterozygous missense NOTCH2 mutation., 2022, 76(7): 1041–1043.
[23] Lu J, Chiang J, Iyer RR, Thompson E, Kaneski CR, Xu DS, Yang CZ, Chen M, Hodes RJ, Lonser RR, Brady RO, Zhuang ZP. Decreased glucocerebrosidase activity in Gaucher disease parallels quantitative enzyme loss due to abnormal interaction with TCP1 and c-Cbl., 2010, 107(50): 21665–21670.
[24] Pagac M, Cooper DE, Qi YF, Lukmantara IE, Mak HY, Wu ZY, Tian Y, Liu ZH, Lei M, Du XM, Ferguson C, Kotevski D, Sadowski P, Chen WQ, Boroda S, Harris TE, Liu G, Parton RG, Huang X, Coleman RA, Yang HY. SEIPIN regulates lipid droplet expansion and adipocyte development by modulating the activity of glycerol-3- phosphate acyltransferase., 2016, 17(6): 1546–1559.
[25] Pipalia NH, Cosner CC, Huang A, Chatterjee A, Bourbon P, Farley N, Helquist P, Wiest O, Maxfield FR. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts., 2011, 108(14): 5620–5625.
[26] Munkacsi AB, Chen FW, Brinkman MA, Higaki K, Gutiérrez GD, Chaudhari J, Layer JV, Tong A, Bard M, Boone C, Ioannou YA, Sturley SL. An “exacerbate- reverse” strategy in yeast identifies histone deacetylase inhibition as a correction for cholesterol and sphingolipid transport defects in human Niemann-Pick type C disease., 2011, 286(27): 23842–23851.
[27] Hutt DM, Herman D, Rodrigues APC, Noel S, Pilewski JM, Matteson J, Hoch B, Kellner W, Kelly JW, Schmidt A, Thomas PJ, Matsumura Y, Skach WR, Gentzsch M, Riordan JR, Sorscher EJ, Okiyoneda T, Yates JR, Lukacs GL, Frizzell RA, Manning G, Gottesfeld JM, Balch WE. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis., 2010, 6(1): 25–33.
[28] Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Görgün CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes., 2006, 313(5790): 1137–1140.
[29] Jin J, Cao LF, Zhao ZH, Shen SX, Kiess W, Zhi DJ, Ye R, Cheng RQ, Chen L, Yang Y, Luo FH. Novelgene mutation E189X in Chinese congenital generalized lipodystrophy child with early onset diabetes mellitus., 2007, 157(6): 783–787.
[30] Antuna-Puente B, Boutet E, Vigouroux C, Lascols O, Slama L, Caron-Debarle M, Khallouf E, Lévy-Marchal C, Capeau J, Bastard JP, Magré J. Higher adiponectin levels in patients with Berardinelli-Seip congenital lipodystrophy due to seipin as compared with 1-acylglycerol-3-phosphate- o-acyltransferase-2 deficiency., 2010, 95(3): 1463–1468.
[31] Nishiyama A, Yagi M, Awano H, Okizuka Y, Maeda T, Yoshida S, Takeshima Y, Matsuo M. Two Japanese infants with congenital generalized lipodystrophy due tomutations., 2009, 51(6): 775–779.
[32] Simha V, Garg A. Inherited lipodystrophies and hypertriglyceridemia., 2009, 20(4): 300–308.
[33] Patni N, Garg A. Congenital generalized lipodystrophies—new insights into metabolic dysfunction., 2015, 11(9): 522–534.
[34] Musso C, Cochran E, Javor E, Young J, Depaoli AM, Gorden P. The long-term effect of recombinant methionyl human leptin therapy on hyperandrogenism and menstrual function in female and pituitary function in male and female hypoleptinemic lipodystrophic patients., 2005, 54(2): 255–263.
[35] Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, Wagner AJ, DePaoli AM, Reitman ML, Taylor SI, Gorden P, Garg A. Leptin-replacement therapy for lipodystrophy., 2002, 346(8): 570–578.
[36] Mulligan K, Khatami H, Schwarz JM, Sakkas GK, DePaoli AM, Tai VW, Wen MJ, Lee GA, Grunfeld C, Schambelan M. The effects of recombinant human leptin on visceral fat, dyslipidemia, and insulin resistance in patients with human immunodeficiency virus-associated lipoatrophy and hypoleptinemia., 2009, 94(4): 1137–1144.
[37] Simha V, Subramanyam L, Szczepaniak L, Quittner C, Adams-Huet B, Snell P, Garg A. Comparison of efficacy and safety of leptin replacement therapy in moderately and severely hypoleptinemic patients with familial partial lipodystrophy of the Dunnigan variety., 2012, 97(3): 785–792.
[38] Scroggins BT, Robzyk K, Wang DX, Marcu MG, Tsutsumi S, Beebe K, Cotter RJ, Felts S, Toft D, Karnitz L, Rosen N, Neckers L. An acetylation site in the middle domain of Hsp90 regulates chaperone function., 2007, 25(1): 151–159.
[39] Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug., 2007, 25(1): 84–90.
[40] Lu J, Yang CZ, Chen M, Ye DY, Lonser RR, Brady RO, Zhuang ZP. Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease., 2011, 108(52): 21200–21205.
[41] Pipalia NH, Subramanian K, Mao S, Ralph H, Hutt DM, Scott SM, Balch WE, Maxfield FR. Histone deacetylase inhibitors correct the cholesterol storage defect in most Niemann-Pick C1 mutant cells., 2017, 58(4): 695–708.
[42] Ramalingam SS, Parise RA, Ramanathan RK, Lagattuta TF, Musguire LA, Stoller RG, Potter DM, Argiris AE, Zwiebel JA, Egorin MJ, Belani CP. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies., 2007, 13(12): 3605–3610.
A study of congenital generalized lipodystrophy (CGL) caused bygene mutation
Jingya Ye1, Aijie Huang1, Zhenzhen Fu1, Yingyun Gong1, Hongyuan Yang2, Hongwen Zhou1
Congenital generalized lipodystrophy (CGL) is an extremely rare genetic disease mainly characterized by absence of whole-body adipose tissue and metabolic dysfunctions such as insulin resistance, diabetes mellitus, hypertriglyceridemia, hepatic steatosis, and acanthosis nigricans. In this study, we reported a novel case of a young woman patient with CGL. The patient came to the hospital for early-onset lipodystrophy and diabetes. She was 19-year-old with a height of 160 cm, a weight of 46 kg, BMI of 17.9 kg/m2, and a serum leptin level of 0.14 μg/L. Genomic DNA was extracted from blood samples of the patient and her family members, including her mother, father and brother. Genetic analysis revealed compound heterozygous mutations of thegene (c.560A>G and c.565G>T) in the patient. Her father carried a heterozygous mutation (c.565G>T), and her mother carried a heterozygous mutation (c.560A>G) in thegene. The mutant p.Y187C plasmid was transfected into HEK293T cells. The protein expression of SEIPIN and its interaction with glycerol-3-phosphate acyltransferase (GPAT3) were observed to be reduced. In addition, based on primary cultured skin fibroblasts from the patient, SEIPIN protein was decreased, and lipid droplets were much smaller when fatty acid was stimulated compared with those observed from healthy subject controls. However, histone deacetylase inhibitors (HDACis) was found capable of rescuing SEIPIN protein in fibroblasts of the patient. In addition, we further summarized and discussed gene mutations ofreported in the current literature. Collectively, these findings have expanded the clinical phenotype and pathogenic gene spectrum of CGL, which might help clinicians to achieve better management of lipodystrophy.
CGL;/SEIPIN; fibroblast; HDACi; diabetes
2022-06-30;
2022-09-12;
2022-09-26
国家重点研发计划(编号:2018YFA0506904,2019YFA0802701)和国家自然科学基金项目(编号:82170882,91854122)资助[Supported by the National Key Research and Development Program(Nos. 2018YFA0506904,2019YFA0802701), and the National Natural Science Foundation of China (Nos. 82170882, 91854122)]
叶静雅,博士,医师,研究方向:内分泌与代谢病。E-mail: yezi88999@163.com
周红文,博士,主任医师,教授,研究方向:内分泌与代谢病。E-mail: drhongwenzhou@njmu.edu.cn
10.16288/j.yczz.22-222
(责任编委: 孟卓贤)
猜你喜欢
癌症进展(2022年18期)2022-11-26
井冈山大学学报(自然科学版)(2022年1期)2022-02-28
眼科新进展(2021年12期)2022-01-15
实用老年医学(2021年10期)2021-12-05
云南医药(2021年3期)2021-07-21
药学研究(2021年3期)2021-04-20
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
医学研究杂志(2015年12期)2015-06-10
商情(2009年17期)2009-09-23
