
红花变豆菜叶绿体基因组组装与序列特征分析研究
2022-11-18 02:46任伟超张美琦刘秀波
中草药 2022年22期
王 震,柳 驰,任伟超,张美琦,刘秀波*,马 伟, 3, 4*
• 药材与资源 •
红花变豆菜叶绿体基因组组装与序列特征分析研究
王 震1,柳 驰2#,任伟超1,张美琦1,刘秀波1*,马 伟1, 3, 4*
1. 黑龙江中医药大学药学院,黑龙江 哈尔滨 150040 2. 开姆尼茨工业大学信息学院,德国 开姆尼茨 09111 3. 江苏康缘药业股份有限公司,江苏 连云港 222001 4. 中药制药过程新技术国家重点实验室,江苏 连云港 222001
以红花变豆菜新鲜叶片为试验材料,通过高通量测序手段,对其叶绿体基因组进行组装和序列特征分析,为进一步开展变豆菜属植物的进化和遗传发育提供指导方法。实验流程按照Illumina公司提供的标准操作流程执行,包括样品质量检测、文库构建、文库质量检测和文库测序等流程,使用已报道的变豆菜的叶绿体基因组作为参考序列,得到红花变豆菜完整的叶绿体基因组序列并对其进行组装、特征序列分析和进化分析。红花变豆菜叶绿体基因组具有典型的环状四分体结构,长度155 700 bp,包括1个大的单拷贝区(large single copy,LSC,85 979 bp),1个小的单拷贝区(small single copy,SSC,17 053 bp),1对相反的序列(inverted repeats sequence,IR,26 333 bp);共注释到130个基因,其中蛋白编码的基因86个,tRNA基因36个,rRNA基因8个,AT含量占叶绿体基因组的61.83%;共检测到1个正向长重复序列(forward repeat sequence),3个回文长重复序列(palindromic repeat sequence);此外,还检测到了168个简单重复序列(simple sequence repeats,SSR)位点;与蛋白编码基因对应的密码子偏好使用A/T碱基,编码异亮氨酸的密码子(I)使用次数最高,编码半胱氨酸的密码子(C)使用次数最低;最后与7种已报道的伞形科植物叶绿体基因组和2个外类群物种通过最大似然法(maximum likelihood,ML)构建系统发育树,发现红花变豆菜与变豆菜和直刺变豆菜亲缘关系最为密切。红花变豆菜的叶绿体基因组数据为伞形科植物研究提供了更为详细完善的资料,为该物种叶绿体基因工程和系统进化分析提供参考依据。
红花变豆菜;叶绿体基因组;高通量测序;系统进化;最大似然法
红花变豆菜Fr. Schmidt是伞形科(Apiaceae)变豆菜属多年生草本植物,是一种可食用的山野菜,具有一定的药用价值,变豆菜属全世界约40余种[1]。其生长在山间林下,阴湿及腐殖质较多的地方,海拔200~470 m,主要产地在我国的东北地区,另外,蒙古、朝鲜、日本北部也有分布[2]。变豆菜属植物在中国约有18种,具有一定的经济价值和药用价值[3],薄片变豆菜Hance又叫血经草,具有治疗跌打损伤和风寒感冒等功效[4],天蓝变豆菜Franch.、直刺变豆菜S. Moor和川滇变豆菜Wolff ex Kretsch均为有名的地方药材,具有活血化瘀和止咳化痰的药效,同属的红花变豆菜也可能会具有相应的药用价值[5]。变豆菜属植物主要基于传统的形态学进行鉴定,如从花粉形态[6]和果实微形态出发进行分类,但是部分品种只从形态学鉴定很难区分,导致其传统分类发展缓慢,因此需要寻找新的方法来对其鉴定手段进行补充。
叶绿体普遍存在于陆地植物、藻类和部分原生生物当中,是绿色植物进行光合作用的细胞器[7],功能包括产生色素、合成糖和某些氨基酸等[8]。被子植物中,叶绿体基因组相对保守,绝大多数为双链环状结构,包括1个小的单拷贝区(small single copy,SSC)、1个大的单拷贝区(large single copy,LSC)以及2个编码相同,方向相反的序列(inverted repeats sequence,IR)IRa和IRb[9]。相对于核基因组,叶绿体基因组具有结构保守、碱基变异速率适中、易于测序等多种优势,已广泛应用于各植物类群的系统进化研究[10]。基于本课题组前期对红花变豆菜叶绿体基因组的研究[11],对其序列数据进一步挖掘分析,对组装后的叶绿体基因组结构、长重复序列分析、简单重复序列(simple sequence repeat,SSR)位点、密码子偏好性以及与其他伞形科植物使用最大似然法(maximum likelihood,ML)构建系统发育树,分析近缘物种亲缘关系,为红花变豆菜的鉴定、系统发育研究提供新的方法。
1 材料与方法
1.1 材料
新鲜的红花变豆菜样本于2020年8月20日采集自中国黑龙江省伊春市(N47°81′08′′,E128°90′97′′)。样品保存在黑龙江中医药大学,样品标本号为YCL20190507007,由黑龙江中医药大学马伟研究员鉴定为伞形科变豆菜属植物红花变豆菜Fr. Schmidt。将采集的红花变豆菜新鲜幼嫩的叶片,通过液氮速冻,并进行研磨,于−80 ℃冰箱保存备用。
1.2 方法
1.2.1 DNA提取 使用CTAB法提取红花变豆菜的全基因组DNA。
1.2.2 测序 总DNA样品经武汉贝纳科技服务有限公司检测合格后,用机械打断的方法(超声波)将DNA片段化,然后对片段化的DNA进行片段纯化、末端修复、3′端加A、连接测序接头,使用琼脂糖凝胶电泳进行片段大小选择,进行PCR扩增形成测序文(NEBNext®Ultra™ DNA Library Prep Kit for Illumina®),建好的文库先进行文库质检,质检合格的文库用Illumina NovaSeq进行测序,对测序得到的序列,通过SOAPnuke(version:1.3.0)软件进行低质量数据过滤,过滤标准为去除N碱基含量超过5%的reads、去除低质量(质量值≤5)碱基数目达到50%的reads和去除有adapter污染的reads,最终得到有效数据(clean reads)。
1.2.3 叶绿体基因组组装 使用SPAdes(version:3.13.0;参数:−k 127)[12]软件进行基因组拼接,选用NCBI数据库中近缘参考植物变豆菜叶绿体基因组(MK208987.1)与拼接结果进blastn(version:BLAST 2.2.30+;参数:−evalue 1×10−5)比对,后续使用Gapcloser(Version:1.12)对gap(含N序列)进行补洞。
1.2.4 叶绿体基因组特征分析 利用专门针对叶绿体的注释软件CPGAVAS2(http://47.96.249.172: 16019/analyzer/annotate)[13]进行基因注释并绘图红花变豆菜叶绿体基因组物理图;使用VMATCH(http://www.vmatch.de/)软件(参数:minimal repeat size 30 bp)查找叶绿体基因组中的散在长重复序列片段;采用MISA软件[版本:1.0;默认参数;对应的各个重复单元(unit size)的最少重复次数分别为1-8、2-4、3-4、4-3、5-3、6-3]对叶绿体进行SSR检测;使用CodonW(Version:1.4.4)对密码子偏好性进行分析,统计估算相对同义密码子的使用频率;通过NCBI数据库下载6种伞形科植物,包括变豆菜L.(MK208987)、明日叶L.(MW125613)、隔山香L.(MT501096)、辽藁本L.(MN652885)、防风L.(MN857472)、白芷L.(KT963037)以及2种非伞形科植物玉米L.(NC001666)和拟南芥L.(NC000932)的叶绿体基因组序列,使用MAFFT[14]软件进行多序列比对,最后使用MEGA X[14]软件基于ML法构建系统发育树。
2 结果与分析
2.1 红花变豆菜叶绿体基因组测序及结构解析
基于课题组已报道的研究发现,红花变豆菜的叶绿体基因组大小为155 700 bp,具有典型的环状四分体结构:包括1个LCS区(85 979 bp),1个SCC区(17 053 bp)和2个反向互补的IRs区(26 333 bp),GC含量为61.83%[14]。
后续通过对红花变豆菜叶绿体基因组物理图谱的绘制(图1)及进一步统计分析发现,测序有效数据Q20值为97.41%、Q30值为92.84%,根据比对覆盖度发现变豆菜(MK208987.1)为最优参考序列,其覆盖度为96.44%,不含有gap。IRs、LSC和SSC 4个区域的GC值都存在一定的差异,IRs区域的CG值最高,为42.92%;SSC区域的CG值最低,为32.56%,LCS区域的CG值介于二者之间,为36.39%(表1)。

叶绿体基因组图谱上4个环。从中心向外,第1圈显示红弧和绿弧分别连接的正向重复和反向重复;第2圈显示用短杠标记的串联重复;第3圈为简单重复序列SSR;第4圈显示质体上的基因结构;基因的颜色是根据他们的功能分类来区分
2.2 叶绿体基因组结构基本特征分析
基于课题组已报道的研究发现,红花变豆菜叶绿体基因组共成功注释了130个基因,其中蛋白编码的基因86个,tRNA基因36个,rRNA基因8个[11]。
通过对以上结果进一步的统计分析发现,根据其基因编码功能可以分为4类(表2),第1类是与自我复制有关的基因,分为5个组,分别是tRNA基因、核糖体小亚基、核糖体大亚基、RNA聚合酶亚基和核糖体RNA基因,成员数量分别为26、12、9、4和4个;第2类为与光合作用相关的基因,细分为7组,分别是光系统I、光系统II、ATP合成亚基、细胞色素亚基、二磷酸核酮糖羧化酶大亚基、NADPH乳酸脱氢酶亚基和依赖ATP蛋白酶P基因亚基,成员数量分别为5、15、6、6、1、11和1个;第3类为其他功能基因,包括成熟酶、外膜蛋白基因、C型细胞色素合成基因和乙酰辅酶A羧化酶亚基各1个;第4类为4个未知功能的基因,还需要进一步研究,以确定其功能。
2.3 长重复序列分析
所有的长重复序列包括3种类型,包括正向重复序列(forward repeat sequence)、回文重复序列(palindromic repeat sequence)和串联重复序列(tandem repeat sequence),这些长重复序列可能具有促进叶绿体基因组重排的功能,并且可以增加其居群遗传多样性[16]。在红花变豆菜叶绿体基因组中共发现了1个正向重复序列,3个回文重复序列,并未发现串联重复序列。其中1个正向重复序列和2个回文重复序列非常短,分别为34、34、30 bp,1个回文重复序列非常长,为26 333 bp,是叶绿体基因组的IR区域。利用这些重复序列,可以为以后开发种群进化标记研究提供基础。
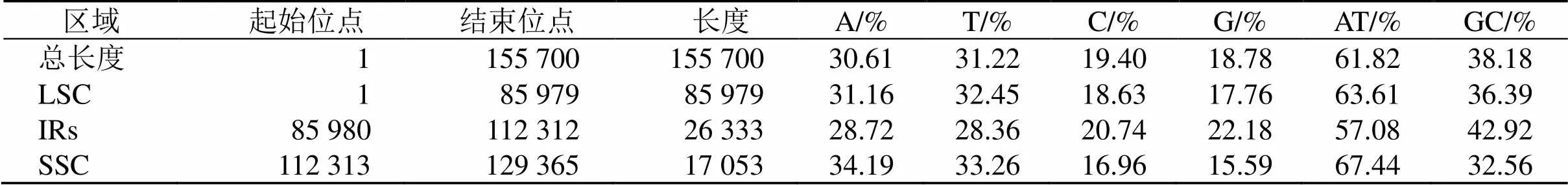
表1 红变豆菜叶绿体基因组碱基组成
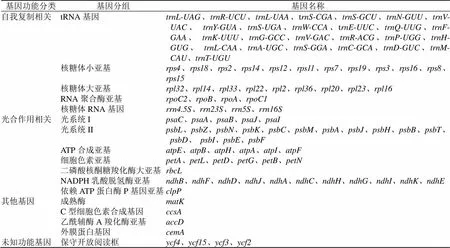
表2 红花变豆菜叶绿体基因组注释基因
2.4 SSR位点分析
SSR又称短串联重复标记、微卫星序列标记[17-18]。SSR表示由基因组中1~6个核苷酸组成的基本单位重复多次构成的一段DNA序列,广泛存在于基因组的各个区域,且侧翼序列通常都是保守性较强的单一序列,可用于个体或物种之间的多态性研究[19]。通过对红花变豆菜叶绿体基因组的分析,共发现了168个SSR位点,分为4个类型,包括复杂重复类型112个,其数量最多;3个碱基重复类型2个,其数量最少;2个碱基重复类型47个;4个碱基重复类型7个,没有单碱基重复类型。97.60%的SSR位点都含有A/T的碱基,仅有4个SSR位点由G/C碱基组成,说明A/T碱基具有碱基偏好性,这可能跟红花变豆菜叶绿体基因组中A/T碱基含量占比高(61.82%)有关联,造成这种偏好性的原因可能与解链难易程度有关。
2.5 密码子偏好性分析
红花变豆菜叶绿体基因组中86个蛋白由21 963个密码子共同编码(表3)。由AUU编码的异亮氨酸数量最多,为876个;由UGC编码的半光氨酸数量最少,仅有60个。3种终止密码子UAA、UAG和UGA在红花变豆菜的叶绿体基因组中使用,其在同义密码子中的占比为50%、25%和25%。通过对86个蛋白编码基因序列分析,得出了同义密码子相对使用度(relative synonymous codon usage,RSCU)[20],RSCU是对同义密码子使用偏好的评估,通过统计发现,由UUA编码的亮氨酸使用最频繁,偏好性最大;由AGC编码的丝氨酸使用频率最低,偏好性最小;由UGG编码的色氨酸和由AUG编码的甲硫氨酸无偏好性。
2.6 红花变豆菜与其他7种伞形科植物叶绿体基因组的比较分析
本研究基于近些年报道的直刺变豆菜[21]、变豆菜[22]、明日叶[23]、隔山香[24]、辽藁本[25]、防风[26]和白芷[27]共7种伞形科植物与红花变豆菜的叶绿体序列长度、编码基因数量及组成结构进行比较(表4)。经过数据统计分析,伞形科内8种植物叶绿体基因组的长度为146 918~155 919 bp,并且都是由典型的四分体结构组成;其大拷贝区和小拷贝区的序列长度差异不大;除了辽藁本的反向重复序列长度为明日叶和防风的2倍,其他7种反向重复序列长度差异也不大;从编码的蛋白质数量来看,红花变豆菜编码的蛋白质数量最多,可能其叶绿体在生长过程中能行使更多的功能。红花变豆菜相比与变豆菜与直刺变豆菜相比,编码的蛋白最多和SSC区序列最长。除此之外,这8种伞形科植物的叶绿体基因组基因顺序和结构与大多数已报道的被子植物叶绿体基因组相似,这说明在植物进化过程中,叶绿体基因序列具有高度的保守性[28]。

表3 红花变豆菜叶绿体基因组密码子使用情况
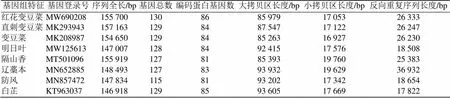
表4 伞形科内8种植物叶绿体基因组特征比较
2.7 叶绿体基因组全序列的聚类分析
叶绿体基因组的聚类分析对植物发育进化研究具有重要意义[28]。通过对红花变豆菜与其他7种伞形科植物叶绿体基因组序列长度,基因结构以及编码蛋白分析,发现其并未有太大差异,于是通过ML法对其构建系统发育树,并加入玉米和拟南芥的叶绿体基因组序列作为外类群,对红花变豆菜进行进化分析,以确定其在伞形科植物中的进化位置(图2)。本课题组发现不同科的植物分为不同的进化分支,8种伞形科植物聚在了1个分支上;说明植物科间关系明确;在这个大的分支基础上,红花变豆菜与变豆菜和直刺变豆菜聚到了1个小分支上,并且自展值为100,说明这3个物种在漫长的进化过程中亲缘关系最为接近。
3 讨论
本研究采用高通量测序技术对红花变豆菜叶绿体基因组进行重测序,并以已报道植物变豆菜叶绿体基因组为参考,成功组装出其完整的叶绿体基因组。相对于传统意义上叶绿体基因组序列的获取,如紫荆泽兰L.[29]和菝葜L.[30]都是采用先从植物样本中分离出叶绿体,然后对其叶绿体进行DNA提取,最终再通过测序技术,实现叶绿体基因组序列的获取,该方法复杂繁琐、不易操作,不利于大范围使用。利用高通量测序技术对红花变豆菜全基因组DNA进行重测序,省略了先分离植物叶绿体再提取叶绿体DNA的复杂操作过程[31],只需提取红花变豆菜全基因组DNA,进行高通量测序,选取已报道植物变豆菜叶绿体基因组序列作为参考基因组,将其所测得的全基因组序列与参考叶绿体基因组序列进行BLASTN比对,提取出关联的叶绿体raw reads,使用过滤软件SOAPnuke对reads进行低质量序列过滤得到clean reads,再利用SPAdes软件对这些序列进行组装及优化,最后使用GapCloser软件对组装结果进行补洞,最终得到完整的红花变豆菜叶绿体基因组序列。本研究使用的方法相较于传统方法,大大简化了试验步骤、降低了试验所需的时间、减少了实验成本,并且该方法实用性广,限制条件少,为大量植物测序叶绿体基因组提供了可能。
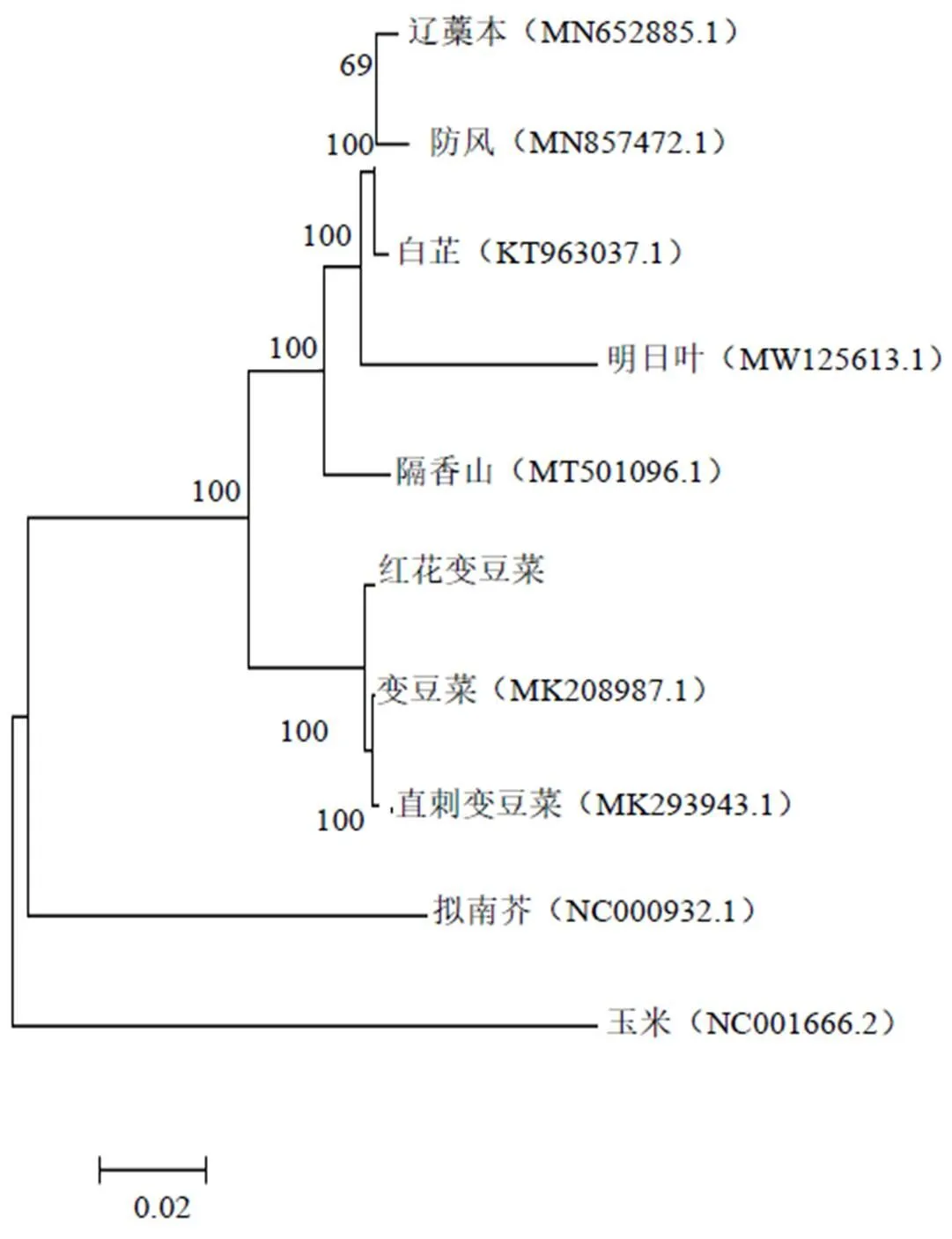
图2 基于叶绿体全基因组10个物种的ML系统发育树结果
变豆菜属L.在克朗奎斯特系统(Cronquist system)、哈钦松系统、恩格勒系统(Engler system)、被子植物发育系统(angiosperm phylogeny group,APG)以及《中国植物志》等各个国家的植物分类体系中都被分为一个自然的类群,归属于伞形科(Apiaceae)、变豆菜亚科(Saniculoideae Drude)[32]。其中一些变豆菜属植物具有一定的药用价值,如变豆菜具有治疗风湿、咳嗽和激活血液循环的功效,直刺变豆菜具有祛风止咳、活血通络和清热解毒的功效,也是一味著名的民族药,收录于《四川省中草药标准(试行稿)》。中国变豆菜属植物约有18种,属于特有的为12种,但是鉴定困难,种间属间易混淆,极其不利于此类植物的发展与利用。针对其分类已经有相关的研究,包括通过形态学研究和分子系统学研究[34]。本研究通过对红花变豆菜叶绿体基本组进行测序及数据挖掘,分析了红花变豆菜的长重复序列、SSR位点和密码子偏好性,为变豆菜属植物研究提供了更为详细、完善的资料, 为该物种叶绿体基因工程和系统进化分析提供参考依据。
利益冲突 所有作者均声明不存在利益冲突
[1] Wyk B E V, Tilney P M, Magee A R. African Apiaceae: A Synopsis of the Apiaceae/umbelliferae of sub-Saharan Africa and Madagascar [M]. Pretoria: Briza Academic Books, 2013: 288.
[2] Vandelook F, Assche J V. Deep complex morphophysiological dormancy in(Apiaceae) fits a recurring pattern of dormancy types in genera with an Arcto-Tertiary distribution [J]., 2008, 86: 1370-1377.
[3] 谢文远, 马丹丹, 陈锋, 等. 黄花变豆菜: 浙江变豆菜属(伞形科)一新种 [J]. 杭州师范大学学报: 自然科学版, 2019, 18(1): 9-12.
[4] 四川省中草药标准(试行稿) [S]. 1979: 10.
[5] 周小江. 肺经草的品质研究[D]. 成都: 成都中医药大学, 2000.
[6] 姚雪莹, 陈志祥, 王奇志. 变豆菜属11种植物的叶表皮微形态特征研究 [J]. 植物研究, 2019, 39(5): 683-691.
[7] 马孟莉, 张薇, 孟衡玲, 等. 豆蔻属药用植物叶绿体基因组密码子偏性分析 [J]. 中草药, 2021, 52(12): 3661-3670.
[8] 邢少辰, Clarke J L. 叶绿体基因组研究进展 [J]. 生物化学与生物物理进展, 2008, 35(1): 21-28.
[9] Zhang T W, Fang Y J, Wang X M,. The complete chloroplast and mitochondrial genome sequences of: Insights into the evolution of plant organellar genomes [J]., 2012, 7(1): e30531.
[10] Daniell H, Lin C S, Yu M,. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering [J]., 2016, 17(1): 134.
[11] Wang Z, Ren W C, Yan S,. Characterization of the complete chloroplast genome ofF. Schmidt ex Maxim [J]., 2021, 6(7): 1999-2000.
[12] Bankevich A, Nurk S, Antipov D,. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing [J]., 2012, 19(5): 455-477.
[13] Shi L C, Chen H M, Jiang M,. CPGAVAS2, an integrated plastome sequence annotator and analyzer [J]., 2019, 47(W1): W65-W73.
[14] Rozewicki J, Li S L, Amada K M,. MAFFT-DASH: Integrated protein sequence and structural alignment [J]., 2019, 47(W1): W5-W10.
[15] Kumar S, Stecher G, Li M,. MEGA X: Molecular evolutionary genetics analysis across computing platforms [J]., 2018, 35(6): 1547-1549.
[16] Tourasse N J, Shtaida N, Khozin-Goldberg I,. The complete mitochondrial genome sequence of the green microalga()reveals a new type of palindromic repetitive repeat [J]., 2015, 16(1): 580.
[17] Farhangian-Kashani S, Azadi A, Khaghani S,. Association analysis and evaluation of genetic diversity in wheat genotypes using SSR markers [J]., 2021, 72(4): 441-452.
[18] Shioi S, Shimamoto A, Nakagami Y,. Precision length determination andsimulation in PCR of microsatellite repeat sequences [J]., 2021, 42(12/13): 1323-1332.
[19] 陈中苏直, 田波, 蔡传涛. 基于SSR分子标记的滇重楼遗传多样性研究 [J]. 中草药, 2017, 48(9): 1834-1838.
[20] 周茜, 陈芸, 王玉州, 等. 独行菜LaBBX基因低温表达响应与密码子偏性分析 [J]. 核农学报, 2021, 35(6): 1253-1262.
[21] Chen Z X, Yao X Y, Downie S R,. Assembling and analysis ofchloroplast genome [J]., 2019, 27(4): 366-372.
[22] Chen Z X, Yao X Y, Wang Q Z. The complete chloroplast genome of[J].DNA Part B, 2019, 4(1): 734-735.
[23] Yan W J, Yang T G, Qin R,. Complete chloroplast genome of(Umbelliferae) [J]., 2021, 6(2): 536-537.
[24] Liao C Y, Gao Q, Wu F. The chloroplast genome of(Apiaceae), an endemic medicinal plant to China [J]., 2020, 5(3): 3782-3783.
[25] Wang S J, Zhang T T, Xu L,. The complete chloroplast genome of(Umbelliferae,L.) [J]., 2020, 5(3): 3025-3027.
[26] Li L L, Geng M L, Li Y S,. Characterization of the complete plastome of(Turcz.) schischk [J]., 2020, 5(1): 786-787.
[27] Zhang R, Xu B H, Cao T Y. Characterization of the complete chloroplast genome of(Apiaceae) as an herb in China [J]., 2020, 5(1): 678-679.
[28] 朱婷婷, 张磊, 陈万生, 等. 1342个植物叶绿体基因组分析 [J]. 基因组学与应用生物学, 2017, 36(10): 4323-4333.
[29] Nie X J, Lv S Z, Zhang Y X,. Complete chloroplast genome sequence of a major invasive species, crofton weed () [J]., 2012, 7(5): e36869.
[30] Liu J, Qi Z C, Zhao Y P,. Complete cpDNA genome sequence ofand phylogenetic placement of Liliales: Influences of gene partitions and taxon sampling [J]., 2012, 64(3): 545-562.
[31] 王虹雨, 李锦萍, 张瑞峰, 等. 基于高通量技术的椭圆叶花锚叶绿体全基因组测序及系统发育分析[J/OL]. 分子植物育种: [2022-10-21]. http://kns.cnki.net/kcms/ detail/46.1068.S.20210611.1420.012.html.
[32] 姚雪莹. 直刺变豆菜的谱系地理学和遗传多样性研究 [D]. 泉州: 华侨大学, 2020.
[33] 陈志祥, 姚雪莹, Stephen R D, 等. 直刺变豆菜叶绿体全基因组及其特征 [J]. 生物多样性, 2019, 27(4): 366-372.
[34] 杨晨, 陈志祥, 姚雪莹, 等. 中国15种变豆菜属植物的花粉形态及系统学分析 [J]. 植物研究, 2020, 40(6): 805-812.
Assembly and sequence analysis of chloroplast genome of
WANG Zhen1, LIU Chi2, REN Wei-chao1, ZHANG Mei-qi1, LIU Xiu-bo1, MA Wei1, 3, 4
1. College of Pharmaceutical Sciences, Heilongjiang University of Chinese Medicine, Harbin 150040, China 2. School of Information, Technical University of Kemnitz, Kemnitz 09111, Germany 3. Jiangsu Kanion Parmaceutical Co., Ltd., Lianyungang 222001, China 4. State Key Laboratory of New-tech for Chinese Medicine Pharmaceutical Process, Lianyungang 222001, China
The chloroplast genome was assembled and the sequence characteristics analyzed with fresh leaves ofas the experimental materials through high-throughput sequencing, so as to provide guidance for the further development of the evolution and genetic development of.The experimental procedures were carried out according to Illumina's standard operating procedures, including the sample quality test, the construction of library, the quality test of the sample library and the sequencing of the library, the reported chloroplast genome ofwas used as a reference sequence, and the complete chloroplast genome ofwas obtained and assembled, and the assembly characteristic sequence analysis and evolutionary analysis were performed.The chloroplast genome ofhad a typical circular quadripartite structure with a size of 155 700 bp in length. In which a large single-copy (LSC) of 85 979 bp and a pair of inverted repeats (IRs) of 26 333 bp were disconnected by a small single-copy (SSC) of 17 053bp. A total of 130 genes were annotated, including 86 protein-coding genes (PCGs), 36 transfer RNA genes (tRNAs) and eight ribosomal RNA genes (rRNAs). AT content accounted for 61.83% of the chloroplast genome. One forward repeat sequence and three palindromic repeat sequences were detected. In addition, 168 simple sequence repeats (SSR) sites were detected. The codon corresponding to the protein coding gene preferred to use A/T base. The codon (I) encoding isoleucine had the highest frequency of use, while the codon (C) encoding cysteine had the lowest frequency. A phylogenetic tree with maximum likelihood (ML) was constructed based on the chloroplast genomes of seven reported Apiaceae species and two other species. It was found thatwere most closely related withand.The chloroplast genome ofprovides more detailed and complete data for the study of Apiaceae plants, and provides a reference basis for chloroplast genetic engineering and systematic evolution analysis of this species.
Fr. Schmidt; complete chloroplast genome; high-throughput sequencing; phylogeny; maximum likelihood
R282.12
A
0253 - 2670(2022)22 - 7183 - 08
10.7501/j.issn.0253-2670.2022.22.022
2022-02-02
中央本级重大增减支项目:名贵中药资源可持续利用能力建设项目(2060302);中药制药过程新技术国家重点实验室开放基金项目(SKL2020M0302)
王 震(1995—),男,硕士,研究方向为药用植物工程。Tel: (0451)87266827 E-mail: 1021707041@qq.com
马 伟,博士,研究员,博士生导师,研究方向为药用植物生物工程与中药资源。Tel: (0451)87266827 E-mail: mawei@hljucm.net
刘秀波(1975—),教授,研究方向为药用植物生物工程。Tel: 13796353268 E-mail: 358270831@qq.com
#共同第一作者:柳 驰(1991—),男,硕士,研究方向为信息分析。Tel: (0451)87266827
[责任编辑 时圣明]
猜你喜欢
快乐语文(2021年34期)2022-01-18
快乐语文(2021年27期)2021-11-24
福建农业学报(2021年6期)2021-08-18
快乐语文(2021年11期)2021-07-20
快乐语文(2021年15期)2021-06-15
课程教育研究·学法教法研究(2019年18期)2019-10-08
少儿科技(2019年9期)2019-09-10
发明与创新·中学生(2019年6期)2019-06-26
生物学教学(2018年2期)2018-08-07
安徽农业科学(2018年1期)2018-05-14
