
遗传性压力易感性周围神经病3例报告及文献复习
2022-11-16 06:49徐文杰李志军毛志娟
中风与神经疾病杂志 2022年10期
李 蕾,徐文杰,聂 清,李志军,毛志娟
遗传性压力易感性周围神经病(hereditary neuropathy with liability to pressure palsies,HNPP)是一种染色体17p11.2的杂合性缺失所致的常染色体显性遗传性疾病,多散发存在,以易受压部位周围神经损害为主要临床表现,临床症状多样,常易漏诊,基因检测可明确诊断。本文将从3个临床病例,结合既往文献报道对HNPP进行讨论。
1 病例资料
患者1,女性,16岁,长期习舞者;因“双下肢麻木无力3 w”入院。患者于3 w前高强度拉腿后出现双下肢麻木无力,麻木主要表现为左小腿外侧、足底及右足底,无明显肌肉萎缩及二便障碍,既往1 y前类似发作,口服甲钴胺后好转。查体:双足背屈肌力4级,弓形足,左腿膝关节以下外侧及前侧、右足背、双足底痛温觉减退,深感觉正常;肌电图示:多发性周围神经源性损害电生理(以髓鞘损害为主)(见表1)。患者血常规、肝肾功能、输血前八项、风湿类风湿免疫全套、甲状腺功能、叶酸、维生素B12、肿瘤全套、骨髓瘤全套均未见明显异常;脑脊液常规、生化、免疫正常范围;血+脑脊液寡克隆带、神经节苷酯抗体、郎飞结抗体、中枢神经脱髓鞘抗体阴性;臂丛神经MRI:双侧颈6、7及右侧颈8臂丛神经稍增粗;腰骶丛MRI:双侧S1、右侧S2神经根起始部点状长T2信号,考虑变性可能;头部MRI、胸部CT、心脏超声无异常;鉴于患者起病年龄较小,双侧起病,既往有类似发作史,需排查遗传性疾病,故给其父母亲行肌电图检查,父亲肌电图未见明显异常,母亲肌电图(见表1);追溯其家族史:患者母亲为普通职工,无长时间过度劳累现象,无肢体麻木、肌力下降等不适,其家族中无类似疾病史。进一步行基因检测示患者及其母亲在基因组:14085603-15472395的位置存在片段大小约1.4 Mb的杂合缺失突变,多重连接探针扩增技术示:PMP22基因外显子1-5杂合缺失,考虑诊断为HNPP。予以营养神经、改善微循环、地塞米松10 mg治疗12 d后患者麻木症状稍好转。出院后继续口服强的松30 mg qd (每5 d减半)、营养神经治疗,停止跳舞。1 m后随访麻木症状较前明显好转,2 y后随访患者诉未再发麻木症状。
患者2,男性,20岁,学生;因“双手麻木10余天”入院。患者10余天前无明显诱因出现双手尺侧麻木,自觉双手小指屈曲稍受限,持物不稳,持续无缓解,无肌萎缩及二便障碍。既往无类似发作史。查体未见明显异常。肌电图示:四肢周围神经源性损害(运动感觉脱髓鞘损害为主)(见表1)。血常规、肝肾功能、输血前三项均未见明显异常;脑脊液常规、生化、免疫正常范围;血+脑脊液神经节苷酯+副肿瘤+郎飞结抗体阴性;臂丛MRI示左侧C5-8神经较右侧稍粗、信号稍高;肺部CT示前纵隔胸腺少许残留;腹部CT示肠系膜淋巴结稍增多;其父亲肌电图(见表1)。患者全外显子检测示:先证者在chr17:14082496-15466816的区域存在大小约1.3 Mb的杂合缺失突变,区域内包括PMP22基因缺失,与基因拷贝数变异测序(copy number variations sequence,CNVSeq)结果基本一致,考虑诊断HNPP。予以营养神经、改善微循环、地塞米松5 mg qd治疗8 d后患者麻木症状稍好转。出院后继续口服强的松20 mg qd (每3 d减半)、营养神经治疗。1 m后随访患者麻木症状较前明显好转,1 y后随访诉出院后仍多次出现双上肢无名指及小指麻木症状,多为提重物或长时间保持肘屈或腕屈后出现。持续2~3 d后自行好转或服用甲钴胺后好转。
患者3,男性,20岁;因“左下肢无力麻木2 m余”入院。患者2 m余前行走较多后出现左下肢无力,左足背屈无力,偶有走路不稳,伴左下肢外侧皮肤麻木,双下肢肌肉疼痛,肉跳,不伴皮温下降及二便障碍。既往儿童时期曾因崴脚出现一次左侧腓总神经损伤;查体:左下肢背屈肌力稍差、双下肢腱反射活跃、左下肢腓侧皮肤浅感觉减退;肌电图示:四肢周围神经源性损害(累及运动感觉髓鞘、神经根)(见表1)。血常规、肝肾功能、输血前八项等均未见明显异常;脑脊液常规、生化、免疫正常范围;血+脑脊液神经节苷酯+郎飞节抗体阴性。臂丛MRI示: C3-6脊髓线样长T2信号影,考虑局限性中央管扩张可能;腰骶丛MRI示:双侧S1、S2、右侧S3神经根周围小囊状长T2信号影,考虑为神经束膜囊肿可能。患者母亲肌电图未见明显异常。父亲及奶奶肌电图(见表1)。患者全外显子检查示:chr17:14096041-15472395的区域存在大小约1.3 Mb的杂合缺失,区域内包括PMP22基因缺失,与CNVSeq结果基本一致,考虑诊断为HNPP。予以营养神经、改善微循环、地塞米松10 mg治疗8 d后患者麻木无力症状较前稍好转。出院后继续予以强的松30 mg qd (每5 d减半)、营养神经治疗。1 m后随访患者诉麻木症状较前明显好转,1 y后随访患者诉麻木症状间断存在,程度较轻,不影响日常生活及运动,未用药。
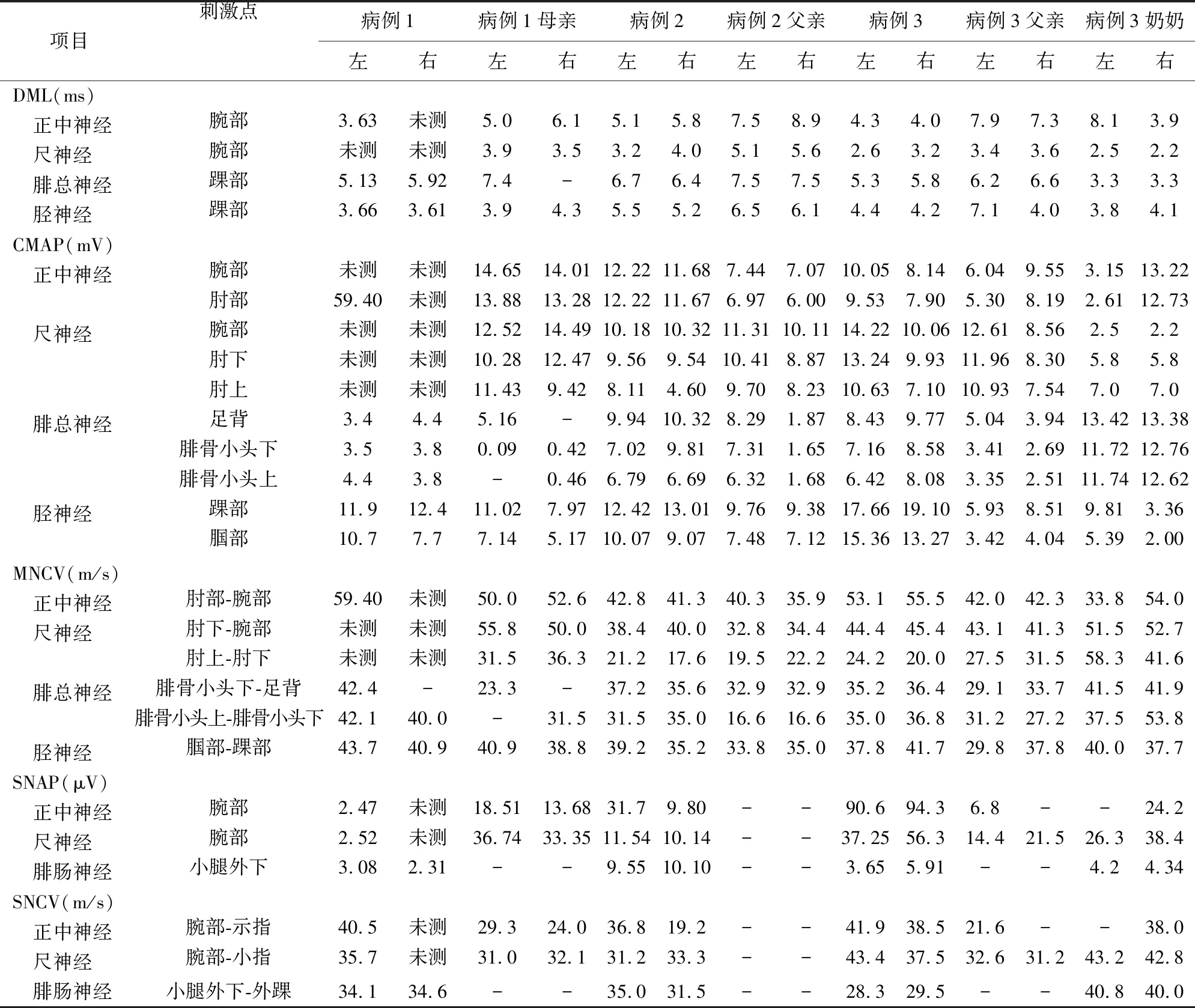
表1 患者及部分家属神经电生理表现
2 讨 论
遗传性压力易感性周围神经病是一种罕见的以周围神经病变为主要表现的常染色体显性遗传病;发病率约为16/100000,其发生率略低于腓骨肌萎缩症(Charcot-Marie-Tooth,CMT)发病率(1/2500~1/10000)[1],发病年龄多在10~30岁,无明显性别差异。超过50%的遗传性周围神经病患者与周围脱髓鞘磷脂蛋白22(peripheral myelin protein 22,PMP22)基因突变有关,包括CMT、 HNPP、DSS、RLS、CMT1E 及 PMP22 突变所致的其他周围神经病等亚型[2];最常见的类型为CMT和HNPP[3]。 PMP22主要在外周神经系统的致密髓鞘中表达,其水平的改变是由基因突变引起的。PMP22蛋白的减少会破坏连接蛋白复合物的形成并产生高渗透性髓鞘,导致失去节间电流和传导阻滞[4],从而导致周围神经病。在绝大多数HNPP患者中,其致病突变是染色体17p11.2的大片段缺失[5],虽然该染色体带有PMP22基因和其他基因,但不足的PMP22单独产生与染色体17p11.2杂合缺失患者相同的表型[6]。这一发现证实了PMP22是该疾病的原因,而不是染色体17p11.2中的其他基因。即包括PMP22基因点突变和包含PMP22基因的缺失突变。有笔者统计发现:86.4%的中国 HNPP 患者为缺失突变,10.2%的患者为点突变[7]。染色体17p11.2的重复则被认为会导致腓骨肌萎缩症[8]。本文中患者3发病年龄在16~20岁,符合HNPP的高发年龄段,其亲代肌电图提示亲代有周围神经损害,基因检测均提示患者3存在染色体17p11.2区含PMP22基因的缺失突变,证实了患者3 HNPP的诊断。
HNPP主要临床表现为反复发作肢体麻木及无力,疼痛较为少见,少部分患者以跛行起病[9]。严重时出现感觉障碍及麻痹症状,症状多出现于受压部位:如腓神经易受压部位在腓骨小头,正中神经在腕管,尺神经在神经沟,并且桡神经、臂丛神经也容易受损[10];也有少部分病例可累及中枢神经[11]。受累神经以腓神经和正中神经、尺神经最为多见,分别占25.0%和21.4%、21.4%,其次是胫神经(18.0%),桡神经(7.1%)和臂丛神经(7.1%)相对少见[12]。本文所述病例1和病例2分别表现有右侧臂丛和左侧臂丛神经增粗,询问病史发现患者均有侧睡习惯,且病例2侧睡后有明显同侧上肢麻木病史。考虑神经增粗可能和患者睡眠习惯有关。类似病因引起的臂丛神经损害也有报吿[13];该病起病时间长短不一,美国曾报道1例与军训相关的爆发型发作,表现为短时间内(3 w)出现右肩无力、疼痛、肌肉萎缩及双上肢麻木无力[14]。HNPP患者特有的电生理表现是感觉神经弥漫性传导速度减慢以及神经卡压部位的运动潜伏期延长[15];少部分伴有轴突损伤[14]。腓骨肌萎缩症(CMT)电生理表现为弥漫性运动、感觉周围神经病变,表现为神经传导速度显著、弥漫性、均匀减慢[16];韩国学者对包括8名HNPP (50个运动神经,39个感觉神经)和6名CMT1A (28个运动神经,16个感觉神经)患者进行临床、电生理、分子学研究发现:97%的HNPP神经感觉神经传导缓慢。其中87.5%常见卡压部位的运动神经传导也异常缓慢,而在非卡压部位传导缓慢并不常见。80%的HNPP神经远端运动潜伏期延长[17]。本文所述3个病例中即使麻木症状较为局限,但均出现四肢不同程度的感觉运动神经损害。由上表可见患者电生理异常主要表现为受损部位及易受压部位运动末端潜伏期延长,运动及感觉神经传导速度减慢,部分受损严重的未引出波形,F波潜伏期延长或正常,CMAP波幅多正常不受影响。本文病例中电生理数值显示患者四肢均有神经源性损害,但患者临床表现不全累及至四肢,部分家属肌电图显示异常但未出现临床症状,这可能与该病慢性病程有关。临床表现为肢体麻木疼痛的疾病非常多,大部分可根据神经电生理检查及脑脊液等检查结果来鉴别。与PMP22突变有关的疾病目前大致分为两类,即遗传性压力易感性周围神经病和腓骨肌萎缩症,临床表现均以肢体麻木、痛温觉改变、肌肉萎缩等周围神经损害为主。HNPP以受压部位受累多见,而腓骨肌萎缩症以双下肢远端开始起病,逐渐向近端蔓延,故临床上若出现非对称性肢体麻木无力、痛温觉减退及肌肉萎缩,尤其是青少年患者,应从临床表现、电生理检查及基因等方面进行诊断机鉴别。
遗传性压力易感性周围神经病属于周围神经性病变,故营养神经、改善微循环及小剂量激素有部分抗炎止痛及营养支持治疗效果,辅之以针灸理疗,能对麻木及疼痛症状有部分缓解作用;对于部分遗留有足下垂患者,应积极使用踝足矫形器辅助治疗,避免遗留畸形,同样也可用腕部或肘部夹板垫固定以保护正中神经和尺神经;部分明显活动受压患者可考虑外科手术治疗(腕管松懈或尺骨减压术)[18]。同时应从诱因上做预防,避免长期的关节压迫姿势。
本文回顾性分析华中科技大学同济医学院附属同济医院收治的3名基因诊断为HNPP患者的临床表现、电生理表现、诊断治疗过程及预后,并进行文献复习,为临床医师对该病的进一步认识提供参考。HNPP临床表现多样,容易漏诊或误诊,所以接诊过程中如出现有或无明显诱因出现易受压部位麻木无力、感觉障碍患者,特别是电生理损害与临床症状体征不相符者应考虑慢性病程的遗传性疾病如HNPP。可以进一步完善基因检测以确诊,营养神经、改善微循环、小剂量激素及针灸理疗可能有部分缓解症状效果,更为完善的病因治疗及基因治疗有待进一步发现。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中国肿瘤临床(2022年14期)2022-08-09
昆明医科大学学报(2022年4期)2022-05-23
中国典型病例大全(2022年11期)2022-05-13
中国听力语言康复科学杂志(2021年6期)2021-12-21
昆明医科大学学报(2021年10期)2021-12-02
健康之家(2021年19期)2021-05-23
现代职业教育·高职高专(2018年8期)2018-05-14
上海医药(2017年2期)2017-03-01
中国实用医药(2016年26期)2016-11-07
