
着色性干皮病继发的面部恶性雀斑样痣、恶性黑色素瘤和基底细胞癌1例
2022-11-15 11:50:44喻光莲寸玥婷代子佳刘彤云
皮肤病与性病 2022年5期
喻光莲,寸玥婷,代子佳,王 莹,刘彤云
(1.昆明医科大学第一附属医院皮肤科,云南 昆明 650032;2.南京医科大学第二附属医院皮肤科,江苏 南京210009;3.云南省第三人民医院皮肤科,云南 昆明 650011)
1 临床资料
患者女,30岁。因“面部黑褐色斑疹30年,斑片、斑块20年,肿物3月”于2021年7月来我院就诊。患者自诉半岁时日晒后双侧面部出现红斑,无水疱、糜烂等皮损,逐渐出现散在黑褐色斑疹,无自觉症状,皮损逐渐增多。10岁时左面部出现一个黄豆大小不规则黑色斑片,未予重视及诊治。近5年来左侧面部、右侧面部相继出现多发黑色不规则斑片、斑块,外用中药涂擦后右面颊外侧斑块破溃。3年前双前臂、颈部等曝光部位开始出现色素沉着斑与色素减退斑,无自觉症状,日晒后增多。近3月来右面部内侧一斑片逐渐凸起形成鹌鹑蛋大小肿块,右眼结膜充血,流泪,晨起时分泌物增多。饮食、大小便正常。既往史:既往体健,父母非近亲婚配,家族中均无类似病症。体格检查:一般情况好,右眼结膜充血,无明显视力障碍,心肺腹无明显异常。皮肤科情况:颜面部、颈部、胸前V形区多发密集分布的形状不一、大小不等的色素沉着斑与色素减退斑,部分黑斑融合成片,颜色不均一。双手背、双前臂多发散在针头至米粒大小白色斑点、斑片。右侧鼻翼、右侧面部多个绿豆至蚕豆大小黑褐色斑块、结节,斑块颜色不均一,部分斑块中央凹陷,表面有糜烂、结痂,境界清楚,无压痛。肿块表面光滑,境界欠清,触之较硬,稍隆起,无压痛。左侧面部见蚕豆至甲盖大小的两个黑褐色斑片,颜色不均一,边缘不规则,无糜烂及渗液(图1a~1d)。
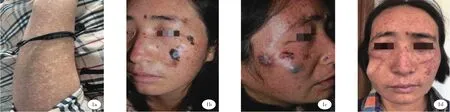
图1 a 前臂多发散在针头至米粒大小白色斑点、斑片;图1b 左侧面部蚕豆至指甲盖大小的黑褐色斑片,斑块,颜色不均一,边缘不规则;图1c 右侧鼻翼、右面部多个绿豆至蚕豆大小黑褐色斑块、结节。图1d 术后3月复查,愈合良好。
实验室检查:尿常规、血常规、粪常规、凝血功能、肝炎病原学、HIV、梅毒、肾功能、肝功能、血糖、电解质、血脂未见明显异常。头颅磁共振未见异常。胸部CT:双肺散在微小结节。术后PET/CT报告提示扫描范围内未见明显异常代谢增高病灶。采用二代测序(NGS)检测黑色素瘤中的10个基因突变情况:TP53、GNA11基因变异,BRAF、CTNNB1、KRAS、MAP2K1、KIT、NRAS、GNAQ、PDGFRA未检出具有明确临床意义的变异。组织病理检查示:(左面颊、右面颊内侧皮损)真表皮交界及上方黑素细胞增生,真皮内可见大小不一的上皮样细胞瘤块,瘤块内可见色素分布(图2a-2b),免疫组化检查:肿瘤细胞Sox-10、HMB-45、Melanin-A:均阳性,P16(部分丢失)、Ki-67(10%-20%+)、ALK(-)。(鼻翼皮损)表皮基底层及其上方黑素细胞增生,部分细胞核大、深染,形态不规则,基底膜完整,真皮浅层散在或灶性炎症细胞浸润,并见噬黑素细胞分布(图2c)。免疫组化检查:Sox-10:基底层及其上方黑色素细胞增生,呈Paget样扩散(图2d)。(右面颊最外侧皮损)基底样细胞组成大小不一团块,境界清楚,瘤块周围细胞呈栅栏状排列,部分瘤块周边可见收缩间隙,可见瘤块向深部侵袭生长(图3)。免疫组化检查:肿瘤细胞Ber-EP4、P53、P40、P63、CKpan、CK5/6: 均 阳 性;Ki-67: 约 20%(+);HMB45、MelanA、Vimentin、S-100:均阴性。

图2 a (HE×40) 真皮内可见上皮样肿瘤细胞构成的瘤块,细胞核大,呈空泡状,核型不规则,核仁明显;图2b (HE×400)2a高倍图;图2c (HE×200) 表皮基底层及上方黑素细胞增生,部分细胞核大,深染,形态不规则,基底膜完整;图2d (envision法×200) 基底层及其上方黑色素细胞Sox-10阳性。
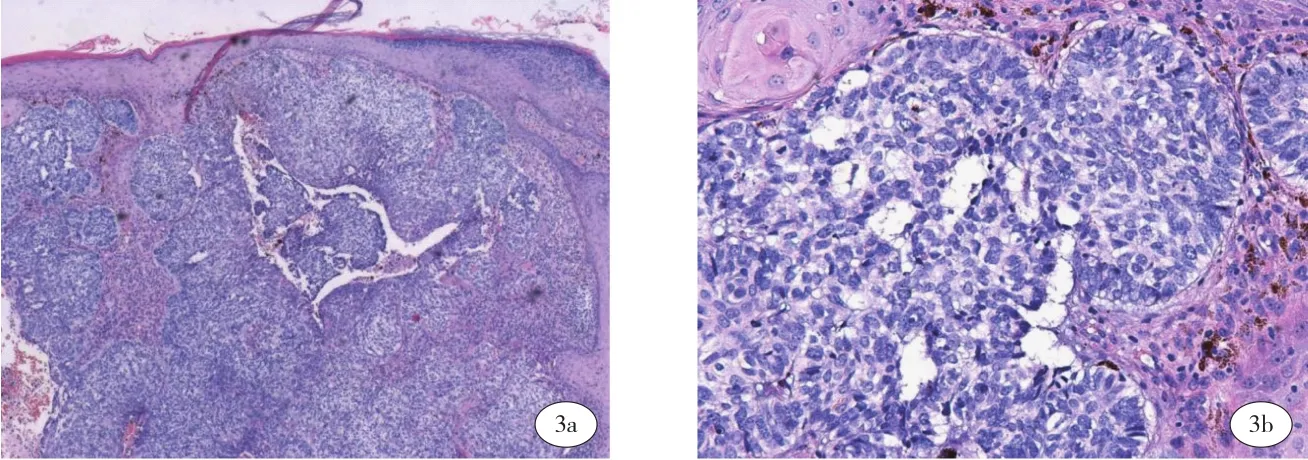
图3 a (HE×40) 真皮内见大小不一的基底样细胞瘤块,境界清楚,瘤块周围细胞呈栅栏状排列,部分瘤块周边可见收缩间隙;图3b (HE×200) 3a高倍图。
诊断:着色性干皮病继发面部恶性雀斑样痣、恶性黑色素瘤和基底细胞癌。
治疗:按照 AJCC第7版皮肤黑色素瘤分期(TNM分期),本例患者分期为T4aN1aM1a,属于Ⅳ期,恶性黑色素瘤浸润深度>1cm(Breslow厚度>4 mm),在全麻下行双侧面部恶性黑色素瘤并基底细胞癌扩大切除术,在病变部位扩大切除2cm左右,术中冷冻切片组织病理检查示四周切缘、基底阴性。由于前哨淋巴结活检阳性及颌下腺查见黑素瘤组织侵及,行功能性颈淋巴结清扫术,将位于颌下三角的颌下腺一起摘除。送检颈部淋巴结20个未查见肿瘤组织转移。由于皮损范围较大,直接缝合容易造成局部组织移位较多、变形明显、疤痕增生,为了让外形修复效果更佳使用了颏下皮瓣进行修复。随访4月至今,原肿瘤部位皮瓣成活,切口愈合良好,形态较好,无创面感染。
2 讨论
着色性干皮病(Xeroderma pigmentosa,XP) 于1874年由皮肤科医生Moriz Kaposi首次描述,是一种罕见的常染色体隐性遗传病,其特征在于皮肤暴露部位的色素变化、紫外线(UV)诱导的皮肤和黏膜癌的风险增加、严重的光敏性和进行性神经变性[1]。XP始于儿童早期,中位年龄为(1~2)岁,在全世界所有种族中均有发生,男女发病率相同[2]。该病由DNA 损伤修复缺陷所致,具有遗传异质性,迄今共有7个互补型和1个变异型。七个XP互补基因(XPA到XPG)负责消除DNA中的紫外线损伤,而第八个(XPV或DNA聚合酶η)负责复制含有未修复损伤的DNA[2]。八个基因中的任何一个基因突变都可以导致XP。由于暴露于紫外线辐射后DNA的核苷酸切除修复缺陷,导致患者对阳光的过度敏感,皮肤经常过早老化,出现干燥,同时含有色素沉着和色素减退区域,并使患者基底细胞癌、鳞状细胞癌和头颈部黑色素瘤等皮肤恶性肿瘤的风险增加1 000倍左右,寿命缩短约30年[3]。眼部受累也很常见,局限于眼前部紫外线暴露的结构(眼睑、角膜和结膜),导致眼睑萎缩、角膜新生血管形成、重度角膜炎、眼部肿瘤(上皮瘤、鳞状细胞癌和黑色素瘤)[4]。20%~30%的患者可出现神经系统疾病,典型的临床表现包括感音神经性耳聋、共济失调、小头畸形、智力障碍、脑肿瘤[5]。神经变性并非在所有类型中均可见,最常与XPA、XPB、XPD、XPF和XPG类型相关,很少与XPC和XPE类型相关[6]。中枢神经系统没有被紫外线直接辐射暴露,发病机制尚不明确,目前认为可能是由氧化代谢诱导的DNA损伤引起的,氧化代谢会杀死神经系统中的非分裂细胞[1,6]。基于对紫外线的极端敏感性,早期曝光部位出现异常皮疹,之后逐渐出现多发的皮肤恶性肿瘤,结合组织病理和免疫组化检查,我们的病例诊断很明确。然而,在一些症状较轻的病例,色素沉着变化直到青春期甚至更晚才出现,此时应注意与红细胞生成性原卟啉、Cockayne综合征、Rothmund-Thompson综合征、Carney综合征、Leopard综合征和Peutz-Jeghers综合征等疾病鉴别。
目前本病尚无满意的治疗措施,因此本病预防大于治疗。头面颈部继发丘疹、溃疡时,应避免应用放疗、化疗、冷冻、电凝等方法,以免因贻误治疗时机而导致肿瘤扩散及转移。彻底切除病灶,创面修复是治疗本病并发症的主要手段[7]。本例患者成功地通过手术切除,用邻近皮瓣转移修复,术后愈合情况良好。
利用NGS技术检测XP恶性肿瘤的基因突变位点,寻找新的治疗靶点是未来XP的一种可能的治疗方法。我们对患者肿瘤组织和血液中循环肿瘤细胞DNA(ctDNA)进行二代(NGS)测序,以试图确定潜在的治疗靶标。我们发现患者GNA11基因的4号外显子(R183)突变率为23.49%,TP53基因4号和5号外显子突变率21.95%、26.33%。GNA11突变与许多癌症有关,常见于葡萄膜黑色素瘤,约占80%~90%,也见于蓝痣和中枢神经系统黑色素瘤,在皮肤黑色素瘤中罕见(<1%),基因突变点位于外显子 5( Q209)和外显子 4(R183)[8]。美国食品药品监督管理局已批准MEK1/MEK2曲美替尼在该特定突变转移性黑色素瘤中的应用[9]。TP53基因位于人类17号染色体的短臂上(17p13.1),有超过50%的肿瘤中存在TP53的基因突变[10]。突变型P53蛋白促进肿瘤细胞增殖、迁移、生存和侵袭,增强肿瘤细胞的耐药性,破坏正常组织的生理结构,促进肿瘤细胞的代谢等[11]。众多临床证据也显示TP53基因突变与多种恶性肿瘤的不良预后有关[12]。然而目前尚无FDA批准靶向TP53基因的抗肿瘤药物。患者由于经济原因,目前尚未采取任何化疗,嘱咐患者做好防晒措施,外出时打伞戴帽,尽量减少外出,必要时皮肤曝光部位涂抹防晒剂,同时严格的防晒措施有可能导致维生素D缺乏,因此应注意补充维生素D。
除此之外,利用下一代测序技术(NGS)检测XP患者基因突变类型将作为分子诊断的重要手段[13]。使用基因疗法和抗氧化剂减少氧化损伤的研究疗法可能是未来的治疗选择[14]。虽然目前XP没有治愈方法,但通过遗传咨询,指导其婚育,可有效降低XP发病率。对于已明确诊断的患者,提高患者意识和关键的早期诊断,以及严格的日光防护和管理,可以显著提高患者的生活质量和预期寿命。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
中国生殖健康(2020年2期)2021-01-18 02:51:26
小学生导刊(2018年13期)2018-06-29 03:49:00
西南国防医药(2016年6期)2016-12-01 06:01:13
癌症进展(2016年10期)2016-03-20 13:15:41
中华皮肤科杂志(2015年8期)2015-11-07 06:08:38
中国麻风皮肤病杂志(2015年9期)2015-10-31 01:43:57
中国麻风皮肤病杂志(2015年9期)2015-10-31 01:43:53
实用手外科杂志(2015年4期)2015-08-27 01:54:26
中国麻风皮肤病杂志(2014年10期)2014-03-29 02:49:14
