
环形RNA全长组装与定量工具的研究进展
2022-11-12 07:54谢宏文宋晓峰
生物信息学 2022年3期
谢宏文,吴 静,2,宋晓峰 *
(1.南京航空航天大学 自动化学院,南京 211106;2.南京医科大学 生物医学工程与信息学院,南京 211166)
环形RNA是一种特殊的内源性非编码RNA,其由前体RNA(Pre-mRNA)经过反向剪接形成,呈封闭环状结构,不具有5’末端帽子和3’末端poly(A)尾巴。研究发现环形RNA参与许多重要的生物学过程,如作为miRNA海绵体,参与基因调控,编码蛋白等。环形RNA的反向剪接位点是鉴别和定量环形RNA的关键,而环形RNA可以通过可变剪接产生不同的环形转录本,这些转录本的全长序列信息以及精确定量对于进一步研究环形RNA功能具有重要作用。生物信息学方法因其能够高效便捷的处理高通量RNA-seq数据,被普遍用来鉴别和分析环形RNA。本文介绍了真核生物环形RNA的产生及功能,对环形RNA检测以及全长组装和定量方面的研究工具进行了综述。
1 环形RNA的产生及其功能
1.1 环形RNA的产生
Sanger等在20世纪70 年代发现某些高等植物中存在可致病的单链环状类病毒,这是人类首次发现环形RNA[1]。但在过去的几十年中,人们把环形RNA看作剪接副产物,环形RNA的研究进展十分缓慢。近年来,随着第二代高通量测序技术的出现及环形RNA分子纯化方法的运用和发展,人们可以重新认识和研究环形RNA。
环形RNA不具有5’末端帽子和3’末端poly(A)尾巴,是以反向剪接的方式形成的一种共价环状结构。环形RNA主要来源于蛋白编码基因,大多由其中的外显子衍生而来并积累在细胞质中,少部分为包含外显子和内含子序列的环形RNA,以及仅来自蛋白编码基因内含子区域的环形RNA[2]。环形RNA的产生机制可分为内含子环化和外显子环化。内含子环化发生在外显子剪接过程中,该过程会产生内含子套索结构的中间产物,这些套索在剪接完成后仍然存在,形成内含子环形RNA(见图1a),如ci-ankrd52由内含子套索结构形成,可以与RNA合成酶Ⅱ结合促进其转录作用[3];外显子环化又可分为三种,即内含子配对驱动环化、套索驱动环化和RNA结合蛋白驱动环化[4]。内含子配对驱动环化将环化外显子的侧翼内含子互补配对,使环化外显子的剪接供体和剪接受体位置更接近,形成环状结构,然后将环状结构中的内含子切除后形成环形RNA(见图1b),如秀丽隐杆线虫侧翼内含子上反向互补重复序列可以使转录本形成发夹结构,促进外显子环化[5];套索驱动环化发生在外显子跳跃过程中,该过程可以使下游5’剪接供体与上游3’剪接受体结合,形成包含外显子的套索结构,再通过套索内的剪接切除内含子,形成环形RNA(见图1c),如Barrett对酵母mrps16基因进行质粒表达,通过删除剪接位点证明了包含外显子的套索结构对于酵母细胞中环形RNA的生成是必要的[6];RNA结合蛋白可以结合到环化外显子两侧的内含子的结合位点上,拉近两端的内含子促进环化,形成环形RNA(见图1d),如可变剪接调控因子quaking可以与环化外显子侧翼内含子上的quaking结合位点结合,促进两侧内含子相互靠近,连接成环[7]。
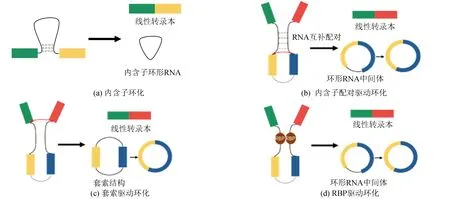
图1 环形RNA环化模型
1.2 环形RNA的功能
由于环形RNA独特的剪接方式及其环状结构,曾经被认为是剪接副产物,不具有生物功能。近年来的研究表明环形RNA广泛存在、结构稳定并且具有组织特异性,在生物体生长发育过程中发挥重要作用[8]。以下将从环形RNA与miRNA相互作用、调控基因表达、与RNA结合蛋白相互作用、翻译蛋白质四个方面介绍环形RNA的功能。
1.2.1 环形RNA与miRNA相互作用
近期研究表明,环形RNA可以作为miRNA分子海绵,通过竞争性的结合miRNA,降低miRNA对其靶分子的抑制作用,进而调控基因表达水平(见图2 a)。Hansen等人研究发现CDR1as(Antisense to the cerebellar degeneration-related protein 1 transcript)含有miR-7的超过70个保守结合位点,可以调节miR-7靶基因的表达[9]。Memczak等人在CDR1as敲除的人类细胞中观察到miR-7靶点的下调,认为CDR1as是作为miRNA海绵来减弱miRNA介导的反应[10]。You等人经过研究认为大多数circRNA并不充当miRNA海绵,并不比其同源mRNA具有更多的miRNA结合位点[11]。
1.2.2 环形RNA与RNA结合蛋白相互作用
研究表明,RNA结合蛋白(RBPs)如Argonaute、RNA聚合酶II和MBL可与环形RNA结合。一些环形RNA可以储存、分类或定位RBP,并且可能像它们调节miRNA活性一样,通过作为竞争元素来调节RBP的功能(见图2a)[12]。环形RNA独特的三级结构在RNA或RBP复合物的组装中起重要作用,其可以作为脚手架(Scaffolding)为蛋白质与RNA、蛋白质与DNA、蛋白质与蛋白质之间的相互作用提供平台[13]。
1.2.3 环形RNA调控基因表达
环形RNA可以认为是一种选择性剪接产物,因此环形RNA可能在选择性剪接水平上起到调节基因表达的作用。环形RNA可与U1 snRNP相互作用,增强其亲本基因的表达(见图2 b)[14]。Burd等人通过研究推测cANRIL(Circular ANRIL)具有转录调控功能,cANRIL的形成降低了ANRIL转录本的表达,从而对编码基因INK4/ARF的转录进行调控[15]。Yang等人发现环形RNA circ-ITCH可以作为海绵体结合miR-17和miR-224,上调靶基因p21和PTEN的表达[16]。
1.2.4 环形RNA翻译蛋白质
环形RNA缺乏帽依赖性翻译的必需元件,例如5'帽子和poly(A)尾,而有些环形RNA也可以翻译产生蛋白(见图2 c)。环形RNA的非帽依赖性翻译可以通过内部核糖体进入位点(IRES)或在5'非翻译区(UTR)中加入m6A修饰后发生[17]。Legnini等人证明circ-ZNF609包含开放阅读框(ORF),可以通过非帽依赖性翻译编码蛋白控制肌细胞的增殖[18]。Yang等人通过Northern blotting和质谱检测等技术发现circ-FBXW7能够编码蛋白FBXW7-185 aa,可以控制癌细胞周期和增殖[19]。
2 环形RNA检测工具
生物信息学方法因其能够高效便捷的处理高通量RNA-seq数据,被普遍用来鉴别和分析环形RNA。为了研究和探索环形RNA的普遍性质和多样功能,研究者们开发了各种计算工具从RNA测序数据中检测环形RNA。这些工具根据检测方法可以为两类:基于注释检测和不基于注释检测。
2.1 基于注释的环形RNA检测工具
基于注释的环形RNA检测工具根据鉴定策略可分为两类,第一类是基于“伪参考序列”的检测策略,第二类是基于“比对片段”的检测策略。
“伪参考序列”检测策略需要根据基因注释信息来构建环形RNA的伪序列,通过伪序列检测环形RNA。基于这种策略的检测工具有KNIFE和PTESFinder等[20-21]。其中KNIFE应用较为广泛,该工具首先从基因组注释信息中构建所有可能的无序“外显子-外显子”连接序列,在Bowtie2的帮助下,将读取的数据分别映射到基因组、rRNA序列、线性“外显子-外显子”连接序列和无序“外显子-外显子”连接序列[22]。真实的BSJ读段必须覆盖指定的核苷酸数量且不能比对到基因组和rRNA序列以及规范的线性“外显子-外显子”连接序列。KNIFE增加了从头分析模块检测来自未注释剪接位点的环形RNA,但Zeng等人认为这种从头检测方法是基于统计学进行推断,不能提供准确的断点[23]。
“比对片段”的检测策略的大致思路便是将测序读段与参考基因组比对,跨越BSJ(Back-spliced junction)的读段被分裂成片段并以相反的顺序与参考基因组对齐,根据这些读段去识别反向剪接位点。基于该策略的工具有CIRCexplorer、UROBORUS和DCC等[24-26]。其中CIRCexplorer使用较为广泛,该工具首先使用TopHat将读段比对到参考基因组,然后提取未映射的读段与TopHat Fusion的结果进行比对,若读段映射到染色体上的顺序相反且映射位置与已知基因注释中的外显子剪接边界一致,便得到环形RNA的反向剪接位点。
相较于不基于注释的检测方法,基于注释的检测方法能够更可靠的检测反向剪接位点,但无法应用于缺乏基因组注释的物种上。
2.2 不基于注释的环形RNA检测工具
不基于注释的环形RNA检测工具有circRNA finder、find_circ和CIRI等[10,27-28]。其中CIRI使用较为广泛,该工具使用BWA-MEM将读段映射到参考基因组[29]。与上述工具需要从未映射上的读段中提取锚序列来检测反向剪接不同,BWA-MEM可以自动确定环形RNA派生读段的断点。CIRI对BWA-MEM比对完成的结果进行两次扫描计算,过滤掉假阳性的反向剪接位点,精度和灵敏度优于其他检测工具,且运算时间耗费不大,实现了更好的平衡性能。相较于基于注释的检测方法,不基于注释的检测方法应用范围更广,能够检测新的候选环状RNA,但需要提高检测和过滤过程中的灵敏度和准确性。
3 环形RNA全长组装与定量工具
研究表明环形RNA在同一个反向剪接位点内部可通过可变剪接形成多个不同的转录本(见图3)[19]。若要深入研究环形RNA的功能特性,必须获取环形RNA转录本内部全长序列信息以及对不同环形RNA内部可变剪接产物进行精确定量。研究者们开发了多种计算工具用于环形RNA内部结构的探索、全长序列的组装及表达量的分析,目前已知的工具(见表1)。
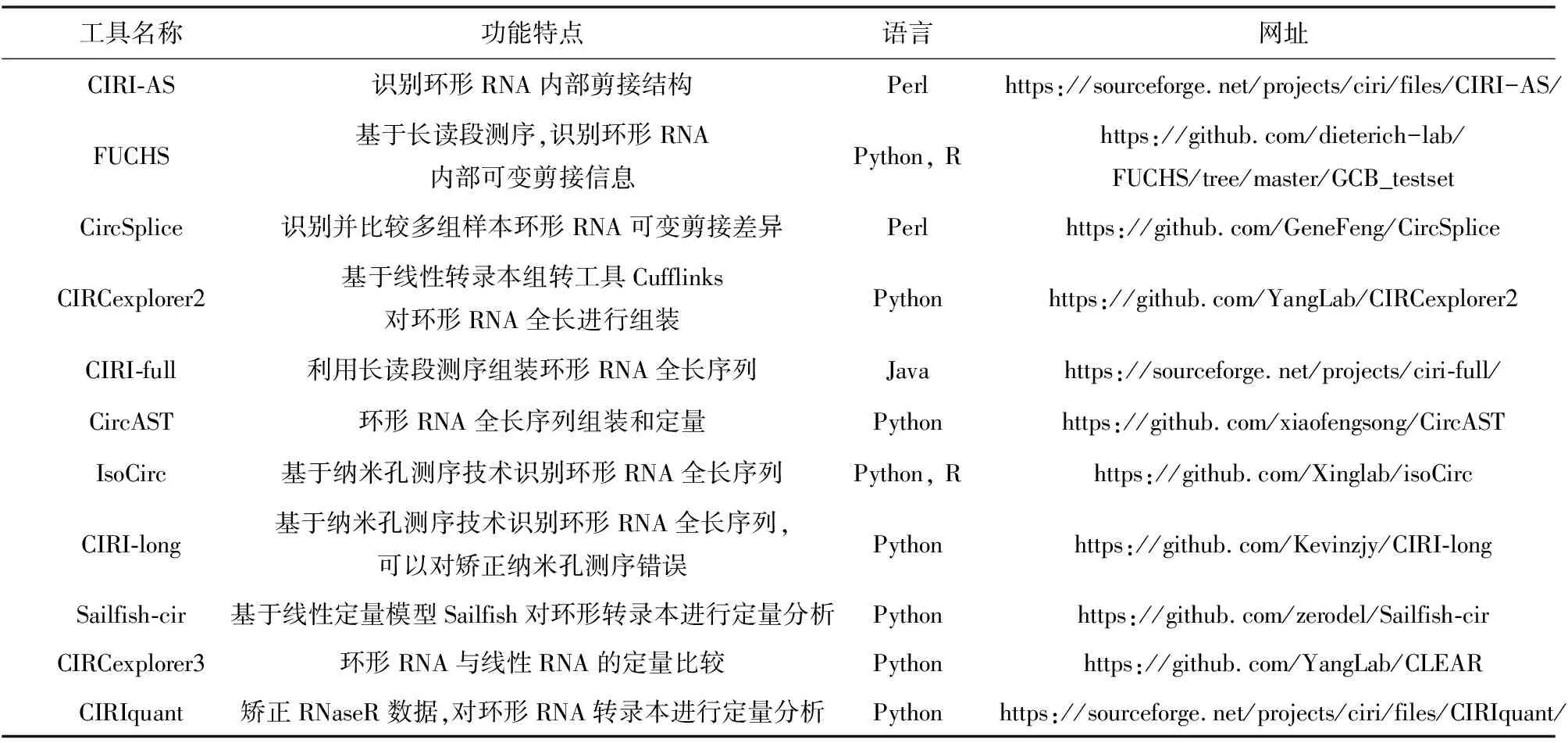
表1 环形RNA下游分析计算工具
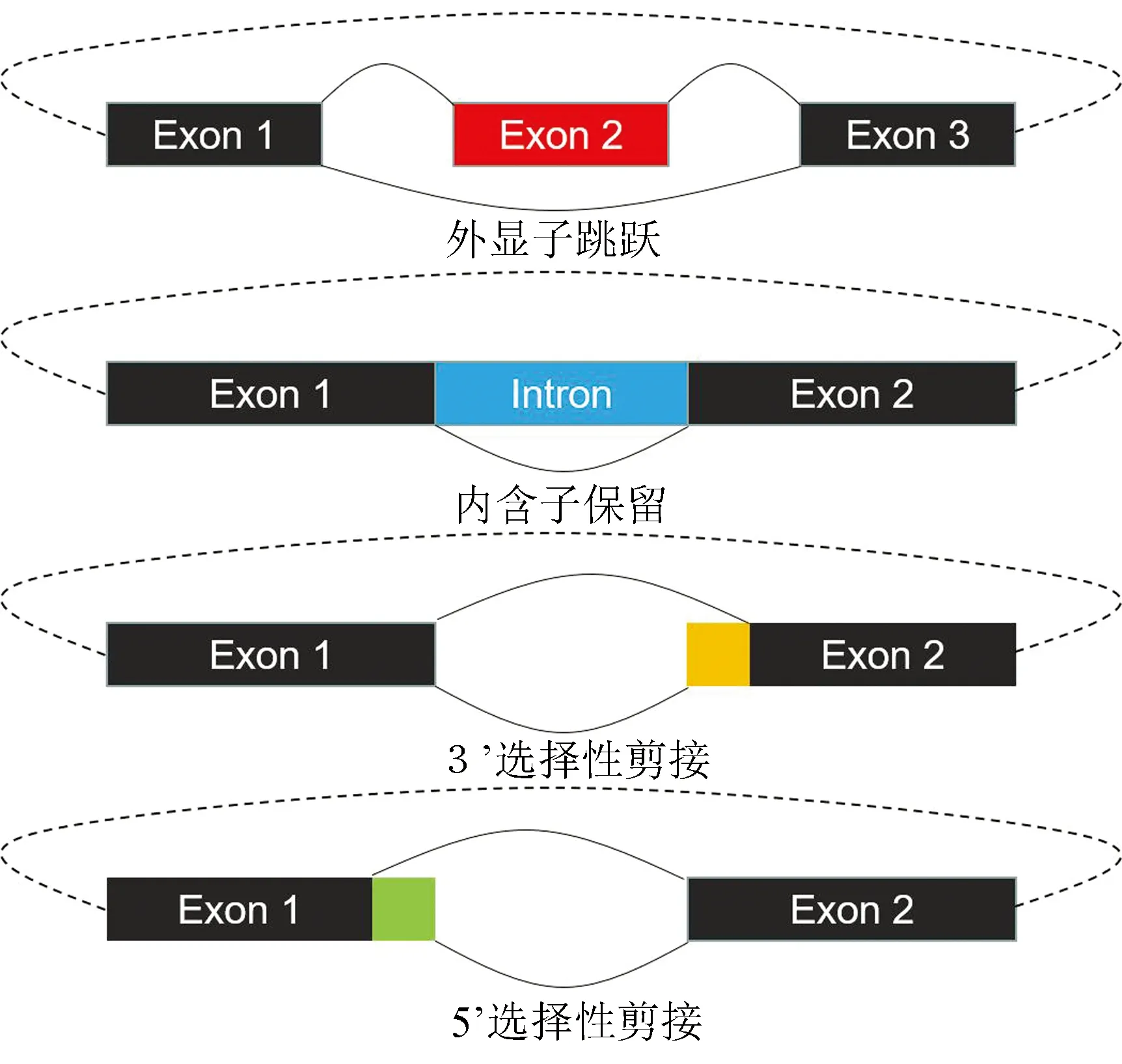
图3 环形RNA内部可变剪接示意图
3.1 环形RNA内部结构探索及全长序列组装工具
为了更好的揭示环形RNA的调控机制,需要深入研究环形RNA内部复杂的选择性剪接结构,构建环形RNA全长序列。目前识别环形RNA内部可变剪接结构的工具主要有CIRI-AS、FUCHS和CircSplice[30-32];针对于环形RNA全长序列组装的工具主要有CIRCexplorer2、CIRI-full、CircAST、isoCirc和CIRI-long[33-37]。
Gao等人开发了CIRI-AS工具研究环形RNA内部的剪接结构[30]。该工具首先利用CIRI找到环形RNA反向剪接位点以及跨越反向剪接位点的读段,然后利用双端测序信息,分析跨越反向剪接读段的另一端读段是否支持环形RNA内部的前向剪接位点,根据支持前向剪接位点读段信息构建前向剪接图,列出可能发生可变剪接的外显子和所有可能的路径,以检测环形RNA内部的可变剪接事件。通过计算与实验验证相结合的手段,揭示了外显子跳跃,5’或3’可变剪接位点以及内含子保留这四种可变剪接事件在环形RNA中普遍存在。CIRI-AS证实了环形RNA内部存在可变剪接事件,但并没有给出环形转录本内部的组成。
Metge等人开发了工具FUCHS对环状RNA内可变剪切等信息进行分析和解读[31]。FUCHS基于长读段测序(大于150 bp),在运行前需要用户手动输入比对和环形RNA位点检测信息。FUCHS从输入的比对信息中提取出嵌合比对的读段信息,识别出同一反向剪接位点内的前向可变剪接事件。类似于CIRI-AS,FUCHS利用双端测序信息来剪接验证环形RNA内部的可变剪接,筛选掉只有一段跨越反向剪接位点的双端测序读段实现对假阳性环形RNA的过滤。
Feng等人开发了CircSplice工具用于识别环形RNA内部的可变剪接事件[32]。该工具通过STAR进行比对,通过GT-AG和CT-AC两种剪切位点过滤并识别环形RNA反向剪接位点,保留两端都落在反向剪接位点之间的读段,根据一端比对到反向剪接位点,另一端比对到反向剪接位点内部的读段对外显子跳跃、内含子保留、5’选择性剪接,3’选择性剪接四种可变剪接类型进行判断,计算支持可变剪接事件的读段数量,最后根据基因组坐标融合不同的可变剪接事件,并比较不同样本间的可变剪接差异。相较于上文提到的CIRI-AS,该方法支持不同样本环形RNA可变剪接差异的比较。
CIRCexplorer2是环形RNA检测软件CIRCexplorer升级版[33]。CIRCexplorer2将Tophat未比对上但映射到Tophat-fusion的读段重新比对到已知和从头组装的注释上,可以检测来自注释或新外显子边界的反向剪接位点;增加了检测环形RNA中的可变剪接事件功能用于确定环形RNA全长,该工具通过比较分析ploy(A)+和poly(A)-的数据集识别环形RNA内部可变剪接事件,重构环形RNA转录本序列。虽然CIRCexplorer2能够通过这种方法给出环形RNA的全长转录本,其使用的工具却是线性转录本的组装工具Cufflinks,会得到错误的环形RNA序列,给计算带来偏差。
Zheng等人开发了工具CIRI-full,提出了一种环形RNA转录本组装的新方法[34]。由于二代测序数据读长较短,限制了研究者们只能定位环形RNA的反向剪接位点,难以获得环形RNA内部的全长结构信息。Zheng等人于是增加测序读长到250 bp,在较长的读长下,若某一双端测序读段来自与环形RNA,两端读段会存在反向重叠区(Reverse overlap,RO),该特征不仅可以识别环形RNA的反向剪接位点,而且可以判断该序列是否覆盖整个环形RNA,从而获取环形RNA的全长序列。
2020年,Wu等人开发了组装环形RNA全长序列的工具CircAST[35]。该工具首先利用Tophat的比对结果找到跨越外显子发生剪接的读段信息,后根据用户通过环形RNA识别工具(如UROBORUS、CIRCexplorer2、CIRI2等)检测到的反向剪接位点信息以及基因组注释文件中的外显子边界信息,对每一个基因位点构建反向剪接事件的有向无环图(Directed acyclic graph,DAG),图中的起点和终点对应环形RNA中的两个发生反向剪接的外显子。相较于传统的线性转录本拼接算法,CircAST若检测到某一基因位点存在超过一个反向剪接事件,则构建多个剪接图,因此可以进行准确拼接和组装。CircAST利用扩展最小路径覆盖算法(Extended minimum path cover,EMPC)结合测序读段信息计算出最优拼接方式,实现环形RNA的全长序列的拼接组装。
Xin等人提出了识别环形RNA全长序列的工具isoCirc[36]。由于短读段测序不能通过实验确定环形RNA全长序列的组成,该工具将数据进行线性RNA消化,并进行滚环扩增,提高低丰度环形RNA的比例,后利用纳米孔长读段测序技术得到包含环形RNA全长序列的长度段测序数据。isoCirc基于上述长读段测序数据,识别出其中重复共有的片段,并将两个相同的重复共有片段连接起来。将连接片段比对到参考基因组以识别环形RNA反向剪接位点和环形RNA内部前向剪接位点信息。通过多重策略(如比对质量,BSJ/FSJ 精确度等)对比对片段进行过滤,之后便得到环形RNA反向剪接位点及全长序列信息。Xin等人使用isoCirc对12种人类组织及人类HEK293细胞系进行测试,一共检测到107 147个环形RNA全长序列,其中包含40 628个长度大于500 nt的环形RNA亚型。isoCirc工具利用纳米孔测序技术,弥补了短读段测序的缺陷,提供了一种研究环形RNA全长序列的新策略。
Zhang等人同样基于纳米孔测序技术,开发了CIRI-long工具[37]。在环形RNA建库过程中,CIRI-long与isoCirc稍有不同,在对数据进行线性RNA消化和环形RNA滚环扩增之后,针对于长度更长cDNA序列加入了片段长度筛选流程。经过纳米孔测序后得到长读段测序数据,利用k-mer匹配方法得到长度段中的重复片段,并使用偏序比对修正测序错误,然后将重复片段比对到参考基因组,结合剪接信号信息,识别反向剪接位点和环形RNA内部正向剪接信息,实现了环形RNA的识别及全长序列的重建。Zhang等人评估了该方法在模拟数据上的效果,并与Illumina测序和实时定量RT-PCR进行了比较,验证了该方法的准确性。
3.2 环形RNA定量工具
为了深入研究环形RNA的调控机制以及不同环形RNA转录本之间的表达差异,需要对环形转录本进行精确定量。目前对于环形RNA进行定量的工具主要有sailfish-cir、CIRCexplorer3、CIRIquant、CIRI-full和CircAST[38-40]。
Li等人开发了工具sailfish-cir对环形RNA的表达量进行计算[38]。先前的研究主要根据检测到的反向剪接读段数量来量化环形RNA的表达,这种检测方法由于反向剪接读段较少,精度较低。因此Li等人将检测到的环形RNA在反向剪接位点处切开,将3’端的序列追加到5’端,构成伪线性转录本,并加入到真实线性转录本集合中构成混合转录本集合。后利用对线性转录本定量的工具sailfish对混合转录本进行定量分析。sailfish-cir基于期望最大化模型,通过迭代将读段分配到不同转录本上,并估计这些转录本的表达量。与基于反向剪接读段计数的直接定量法相比,sailfish-cir与qRT-PCR定量结果有更强的相关性。
Ma等人对CIRCexplorer分析流程进行了升级,开发了环形RNA与线性RNA定量比较的工具CIRCexplorer3-CLEAR[39]。该方法提出了一种定量转录本表达量的参数FPB(Fragments per billion mapped bases),该参数相较于FPKM能够不受测序读段长度的影响。该工具首先利用HISAT2将测序数据比对到参考基因组,利用StringTie对线性转录本进行组装,计算跨越前向剪接位点的测序片段得到线性转录本的表达FPBlinear;然后利用TopHat-Fusion处理未比对上的测序片段,通过计算跨越反向剪接位点片段得到环形转录本的表达量FPBcirc,通过环形转录本表达量与对应线性转录本表达量的比值CIRCscore(FPBcirc/FPBliner)衡量环形RNA的相对表达量。相较于其他基于跨越反向剪接位点读段进行定量的方法,该工具提出了新的定量参数FPB,可以利用CIRCscore对环形RNA进行相对定量,具有更广泛的适应性。
Zhang等人开发了鉴定和定量环形RNA的算法CIRIquant[40]。CIRIquant首先利用CIRI等工具从测序数据中识别环形RNA的反向剪接位点,然后基于环形RNA反向剪接位点构造环形转录本伪参考序列,重新映射读段到伪参考序列。CIRIquant运用这种方法可以更准确的识别环形RNA的反向剪接序列,根据反向剪接读段和前向剪接读段的比例对环形RNA进行定量。考虑到环形RNA建库时RNase R处理存在偏差,CIRIquant结合未经RNase R处理过的数据利用高斯混合模型(Gaussian mixture model,GMM)对RNase R处理数据进行校正,使定量更为准确。
除了这些工具外,上文提到的工具CIRI-full和CircAST不仅可以组装环形转录本全长序列,还可以对环形转录本进行表达量估计。CIRI-full在构建环形RNA的全长后,采用蒙特卡洛方法模拟不同环形RNA剪接产物读段在全长序列上的分布,通过梯度下降法求得最优的表达量组合,实现了对不同环形转录本相对丰度的预测评估,并在模拟和真实数据上验证了该方法的准确性。但该方法受到测序长度的限制,需要更高的测序成本和更先进的测序技术来获得较长读长的测序数据;CircAST在实现环形RNA全长序列组装之后,通过期望最大化(Expectation maximization,EM)算法来估计每个组装的环形RNA转录本的丰度。其在似然函数的构造等方面充分注意到环形RNA的结构特点,在绝对定量和相对定量中均表现出了良好的性能。
4 总结与展望
随着深度测序技术和分子纯化方法的发展,人们对于环形RNA的认识正在逐渐发展。在对环形RNA的研究中,计算方法因其在高通量RNA-seq数据分析中的便利性以及在分析环形RNA表达方面的优势而起着重要的作用。由于环形RNA自身的结构特点和其特有的剪接事件,我们不能简单地用线性RNA的相关工具来解决环形RNA的相关计算,需要开发更多针对于环形RNA的计算工具。为了更好的揭示环形RNA的特殊功能,需要对环形RNA内部全长序列进行准确重建。由于二代测序的读段较短,基于二代测序数据重建环形RNA全长序列难度较大;基于纳米孔长读段测序技术构建环形RNA全长的工具将会是未来研究开发的方向。在环形RNA定量方面,现有的工具或基于线性RNA开发而来,或对于输入数据存在特定要求。而在全转录组测序数据上由于环形RNA与线性RNA重叠部分的干扰,精准定量环形RNA转录本仍然是一个挑战。开发基于全转录组测序数据环形RNA特定的计算工具是目前环形RNA研究中迫切需要解决的问题,这对于进一步研究环形RNA的生物学功能至关重要。随着计算算法、测序技术、基因组学和生物信息学的发展,相信会有更多环形RNA相关计算工具的出现,促进对于环形RNA机制和功能的深入研究。
猜你喜欢
分子催化(2022年1期)2022-11-02
烟草科技(2021年6期)2021-06-24
中国生殖健康(2020年4期)2021-01-18
山地农业生物学报(2020年2期)2020-11-09
现代农业科技(2020年15期)2020-08-16
生物学教学(2018年4期)2018-11-29
电脑知识与技术(2018年19期)2018-11-01
生物学教学(2018年12期)2018-08-15
湖北农业科学(2014年11期)2014-09-10
