
MeNQ/NQ低共熔物形成的微观机制
2022-11-04 02:30:44廖奕玫陈伶媛段晓惠
火炸药学报 2022年5期
廖奕玫,陈伶媛,游 婷,段晓惠
(西南科技大学 环境友好能源材料国家重点实验室,四川 绵阳 621010)
引 言
熔铸炸药是一类在液相载体炸药中加入高能固相主炸药(如RDX、HMX等)进行铸装的混合炸药,在军事上有着广泛的应用[1]。目前最常用的载体炸药是TNT,但TNT自身的缺陷导致该类熔铸炸药在成型、贮存及使用过程中,存在空洞[2]、发脆[3]、感度高[4]、毒性大[5]等问题,因此,绿色高性能非TNT熔铸载体炸药的设计与实现成为含能材料领域的研究热点。而目前最主要的研究方法是以新型含能低共熔物替代 TNT 来获得满足使用要求的熔铸炸药。含能低共熔物的研究始于20世纪初,报道有多种含能低共熔物,如甲基硝基胍(MeNQ)基低共熔物[6]、3,4-二硝基呋咱基氧化呋咱(DNTF)基低共熔物[7]、1,3,3-三硝基氮杂环丁烷(TNAZ) 基低共熔物等[8]。其中,MeNQ基低共熔物被认为是最有希望替代TNT的低共熔物之一[9]。研究表明[10],MeNQ可与硝基胍(NQ)、硝酸铵(AN)和硝酸肼(HN)等形成低共熔物。以MeNQ/AN低共熔物为配方组成研制的载体炸药,已取代TNT用于AFX-453型号航弹中[11]。
目前对含能低共熔物的研究,主要集中在相图绘制、样品制备、以及作为液相载体在熔铸炸药中的配方研制与性能测试,对其中涉及到的一些基础科学问题还不清楚,比如在原子分子层次上的形成机制。药物领域在这方面开展了初步研究,结果指出,同为共结晶产物的共晶、固溶体和低共熔物,其形成均起源于组分间的相互作用和尺寸/形貌间的相互匹配[12-14]。共晶的形成是不同分子间的附着力足够强,能够克服主客体分子的尺寸/形貌失配。固溶体发生在同晶(从属于同一空间群,且相应晶面的两面角极为接近)或同构材料(相应结构单元处于相同的等效点系位置的同晶材料)[15]之间,是客体分子采取间隙或取代的方式、以任何比例进入主体晶格中,同时保留主体的晶格结构。和共晶相比,相同分子间的内聚力增强,不同分子间的附着力减弱,二者处于一个较为平衡的状态;低共熔物发生于非同晶(不属于同一空间群)材料之间,和固溶体相比,不同分子间的附着力进一步减弱,且主客体分子间存在尺寸和形貌上的失配,是体系中不同的固溶体为消除内应力和晶格变形而发生分离重组,也被称为不连续的固溶体。同时通过这些研究,药物科学家发现适用于共晶设计的晶体工程学原则[16-19],可半经验性地推广到低共熔物。因此,含能低共熔物在原子分子层次上的形成机制,可通过构建超分子,从主客体分子的官能团结构、分子间作用力的模式和强弱以及分子的形状等方面进行研究。
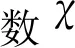
1 计算方法
1.1 第一性原理计算
从剑桥数据库[21]中获取MeNQ[22]和NQ[23]的晶体结构,并从中取出单个分子作为输入文件。由于MeNQ和NQ含多个氢键良给体和受体,分子间可存在多个氢键相互作用,所以结构优化采用了色散校正的密度泛函方法,M06-2X-D3[24]/6-311+G(d, p)[25]。在结构优化的基础上再进行频率计算,以确保其处于稳定结构,并计算得到MeNQ和NQ分子表面静电势。按照氢键供体-受体规则和静电势互补匹配原则构建最可几的二聚体模型。采用和单分子相同的优化方法,对构建的二聚体进行结构优化和频率计算。为了考察不同计算方法对计算结果的影响,对优化得到的最稳定结构再用精度更高的方法,MP2[26]/may-cc-pvtz[27]和PWPB95-D3[28]/ma-def2-QZVPP[29],计算其分子间相互作用能,并采用均衡校正法(Couterpoise Procedure, CP)[30]进行基组重叠误差(BSSE)校正。分子间相互作用能的能量成分分析采用对称匹配微扰理论(Symmetry-Adapted Perturbation Theory,SAPT)[31],解析静电、交换、诱导和色散4种相互作用对作用能的贡献。通过Multiwfn[32]结合VMD[33]软件进行波函数和约化密度梯度(RDG)[34]分析。结构优化、频率计算和MP2单点能计算使用Gaussian16程序[35],PWPB95-D3计算使用ORCA4.2程序[36],SAPT计算在PSI4程序[37]上进行。
1.2 Monte Carlo模拟
2 结果与讨论
2.1 二聚体结构和相互作用能分析
M06-2X-D3/6-311+G(d, p)水平上得到MeNQ和NQ分子的表面静电势如图1所示。对于MeNQ或NQ分子,负电势区域主要分布在硝基氧以及与硝基相连的氮原子周围,而正电势区域则主要分布在氨基氢和甲基氢周围。负电势区域的基团为氢键良受体,正电势区域的基团为氢键良给体。按照氢键形成的基本原则[20],推测MeNQ与NQ的同二聚体和异二聚体可能存在的氢键结构,如C—H…N(O)、N(O)—H…N(O),由此构建最可几的二聚体模型,作为初始猜测进行结构优化。

图1 MeNQ和NQ在0.001a.u.等密度表面的静电势(红色表示正的静电势,蓝色表示负的静电势,色标单位kJ/mol)Fig.1 The electrostatic potentials on 0.001a.u. isodensity surfaces of MeNQ and NQ(The red surface represents the positive part of the electrostatic, and the blue surface represents the negative part of the electrostatic. The color code unit is kJ/mol)
图2列出了在M062X-D3/6-311+G(d, p)水平下相互作用能最低的3种二聚体结构和氢键作用模式。从图2可以看出,MeNQ和NQ分子中氢键给体和受体之间均发生了多氢键作用。由于甲基的引入,除了N—H…O和N—H…N氢键外,在MeNQ-MeNQ和MeNQ-NQ二聚体中还存在强度较弱的C—H…O氢键。3种二聚体的氢键键长在1.906~2.640Å范围内,属于中强氢键(1.2~2.2Å)和弱氢键(3.2~4.0Å)[40]。其中,MeNQ-MeNQ二聚体中的氢键个数最多(8个),其次是MeNQ-NQ(7个),氢键个数最少的是NQ-NQ(6个)。根据氢键个数和键长可初步推测3种二聚体的稳定顺序:MeNQ-MeNQ>MeNQ-NQ>NQ-NQ。在该水平上计算的相互作用能(见表1)也预测了相同的稳定顺序。
除M062X-D3/6-311+G(d, p)方法外,还采用了精度更高的计算方法,MP2/may-cc-pvtz和PWPB95-D3/ ma-def2-QZVPP,基于最稳定的二聚体结构,进行了相互作用能计算,结果列于表1。
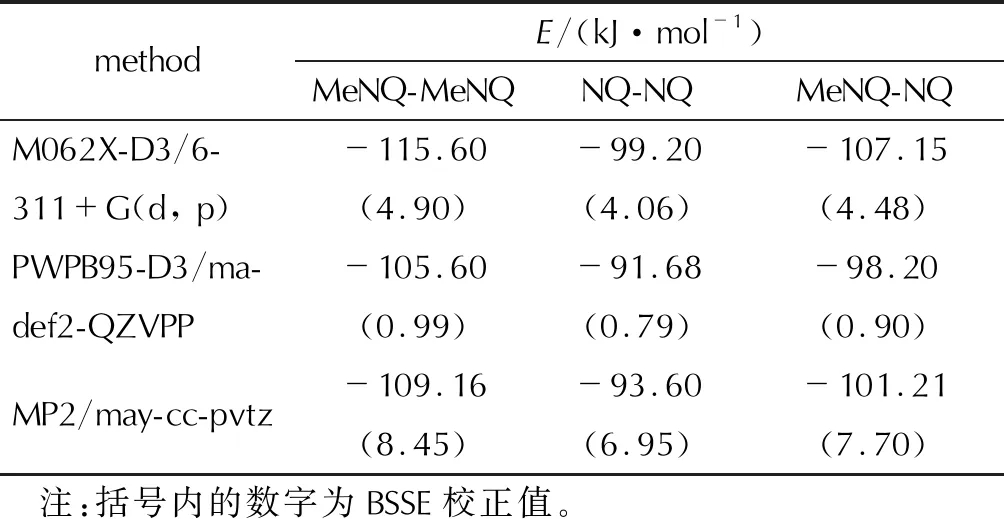
表1 不同方法计算的相互作用能Table 1 Interaction energy calculated by different methods
从表1可以看出,3种方法对二聚体稳定次序的预测完全相同,即MeNQ-MeNQ>MeNQ-NQ>NQ-NQ,和结构分析的预判相一致。M062X-D3/6-311+G(d, p)计算的相互作用能,其绝对值高于MP2/may-cc-pvtz和PWPB95-D3/ ma-def2-QZVPP,说明该方法过分高估了体系中的分子间相互作用。后两种精度更高的计算方法得到的作用能值则非常接近,最大差异为MeNQ-MeNQ的3.56 kJ/mol,约占总相互作用能的3%。PWPB95-D3/ma-def2-QZVPP方法的BSSE校正值最低,对相互作用能的计算影响最小。3种方法对相互作用能的计算结果均说明,在MeNQ与NQ二元体系中,MeNQ分子的内聚力大于其与NQ的附着力而占据支配地位。
图2为3种二聚体形成的多氢键结构,采用对分子间弱相互作用计算精度更高的方法来获得其相互作用能是非常必要的。目前对MeNQ与NQ体系中的分子间相互作用报道很少,仅见李华荣[41]对其进行了研究。结构优化采用MP2/6-31G (d)方法,然后用MP2/6-311+G (d, p)方法计算相互作用能。结果显示其最稳定二聚体结构中的氢键数目均只有两个,远小于图2所示的氢键数目。相互作用能MeNQ-MeNQ:-45.664kJ/mol、MeNQ-NQ:-43.543kJ/mol和NQ-NQ:-42.233kJ/mol,不及表1所列数值的一半,不过预测的相对稳定顺序一致。推测其优化所得的二聚体结构并不是能量最低的,且计算方法的精度有待进一步提高。
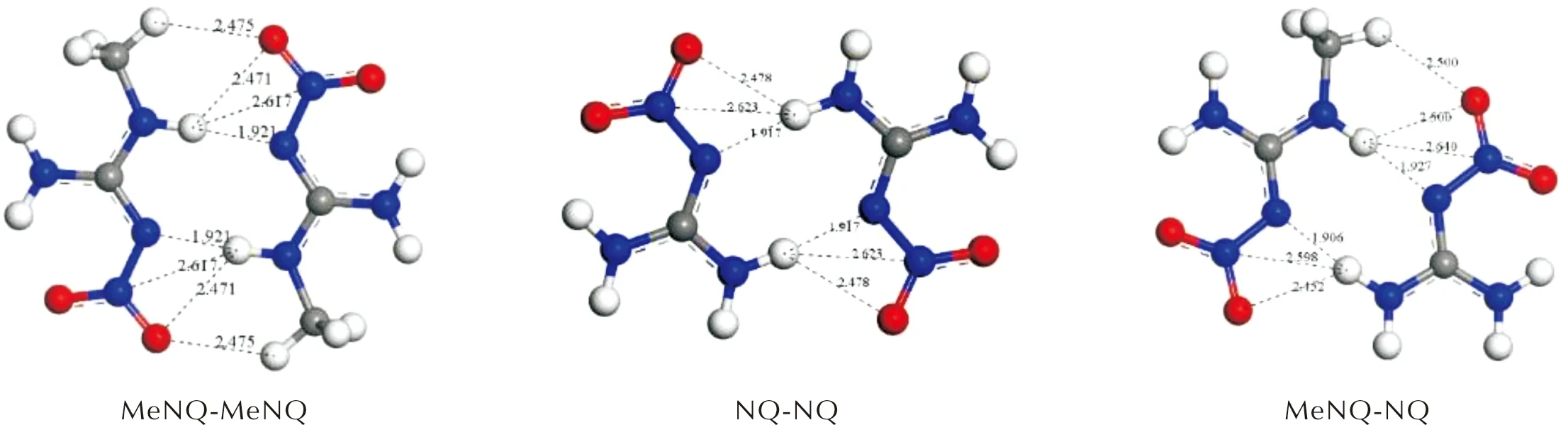
图2 M062X-D3/6-311+G(d, p)水平下3种二聚体的最稳定结构Fig.2 The most stable structures of three dimers at M06-2X-D3/6-311+G(d, p) level
SAPT是一种计算片段间相互作用能的方法[31],常被用于研究弱相互作用,且没有BSSE问题。SAPT将相互作用能(Eint)分解为4部分:静电(Eelest)、交换(Eexch)、色散(Edisp)和诱导(Eind)。本研究采用sSAPT0耦合基组jun-cc-pVDZ进行计算,对3种二聚体相互作用能的分解结果如表2所示。可见分子间相互作用的吸引项主要为静电力,其次为诱导和色散作用(范德华力),Eexch为排斥项。在3种二聚体中,MeNQ-MeNQ二聚体的静电力、范德华力和排斥力均最大,而NQ-NQ中的静电力、范德华力和排斥力均最小。几种力的协同作用导致MeNQ与MeNQ的分子间相互作用最强,MeNQ与NQ次之,而NQ与NQ最弱。
通过RDG分析绘制了3种二聚体的填色等值面图(见图3)[34],图形化地显示了表2中不同种类的作用能发生在哪些基团之间。图中蓝色、绿色和红色分别代表强吸引作用(氢键、卤键等)、范德华作用和强互斥作用(位阻作用)区域。从图3可看出,氢键作用(静电)主要发生在N—H…N—NO2之间(蓝色),排斥作用在N与N以及N与O之间(红色),范德华力则在距离较远的O与H以及H与H之间(绿色)。

表2 3种二聚体在sSAPT0/jun-cc-pVDZ水平下的相互作用能Table 2 The interaction energy components of three dimers at sSAPT0/jun-cc-pVDZ level
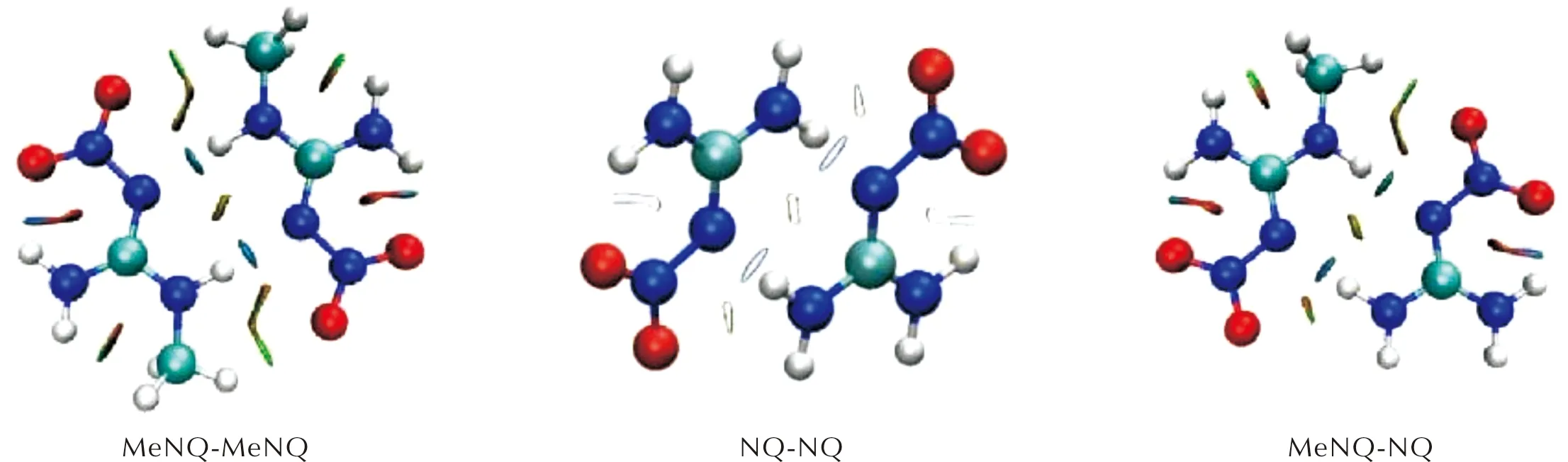
图3 MeNQ-MeNQ、NQ-NQ和NQ-MeNQ 3种二聚体的等值面填色图(蓝-绿-红色标值对应sign(λ2)ρ从-0.05至0.05a.u.的值)Fig.3 The isosurface graph of three dimers for MeNQ-MeNQ, NQ-NQ, NQ-MeNQ (The structures are colored on a blue-green-red scale according to the values of sign (λ2)ρ, ranging from -0.05 to 0.05a.u.)
2.2 分子和晶体结构分析
由上述分子间相互作用能的计算结果可知,对MeNQ和NQ构成的二元体系,MeNQ-MeNQ的分子间作用力大于MeNQ-NQ而占据支配地位。根据 Cherukuvada等[12-14]的研究结果可预测二者无法形成共晶,其共结晶产物可能是低共熔物或固溶体。究竟是低共熔物还是固溶体,取决于组分的晶体和分子结构。
MeNQ和NQ的晶体和分子结构显示在图4中。MeNQ晶体结构属于单斜晶系,P21/N空间群,其晶胞参数为:a=4.632Å,b=10.127Å,c=11.240Å,β=100.164°[22];而NQ晶体结构属于正交晶系,Fdd2空间群,其晶胞参数为a=17.643Å,b=24.883Å,c=3.595Å[23]。可以看出,MeNQ和NQ为非同晶材料。此外,在M06-2X-D3/6-311+G(d, p)水平上优化后的NQ分子呈平面结构,分子体积为68.418cm3/mol。MeNQ是NQ氨基上的一个H原子被甲基取代而成,分子的平面结构遭到破坏,分子体积增大为81.902cm3/mol。因此,MeNQ和NQ分子间存在形貌和尺寸上的失配。
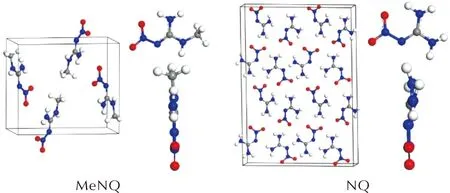
图4 MeNQ和NQ的晶体和分子结构Fig.4 Crystals and molecular structures of MeNQ and NQ
2.3 相互作用参数分析

(1)
(2)
式中:ΔEmix为二元体系的混合能;Zij和Eij分别为分子i和j之间最优的配位数和温度依赖的相互作用能。在作用能和配位数的Monte Carlo模拟过程中,基础分子保持固定,对检测分子随机进行平移和旋转,以得到大量的配对模式及其对应的作用能。能量计算采用分子力场方法,其计算精度对力场的依赖度高。本研究对所采用的Dreiding力场是否适用于MeNQ和NQ体系进行了验证。采用该力场对MeNQ和NQ的单胞进行结构优化,优化结果列于表3。结构优化的参数为:力场指定电荷;“Fine”精度;“Smart”方法;“Ewald”方法计算静电相互作用和范德华力;优化晶胞参数。从表3可以看出,所有误差均小于5%,在分子模拟方法可接受的误差范围之内,说明Dreiding力场可用于MeNQ和NQ体系的分子模拟。

表3 MeNQ和NQ晶胞参数的Dreiding力场模拟值及相对误差Table 3 Dreiding force fieldsimulation values and relative errors of MeNQ and NQ lattice parameters
图5为采样空间中不同配对模式对应的相互作用能分布,可见所有配对模式的作用能分布曲线形态相似,说明MeNQ和NQ可互溶[42],即NQ可溶于MeNQ中形成固溶体,MeNQ也可溶于NQ中形成另一固溶体。体系中这两种固溶体为了消除内应力和晶格变形而发生分离重组,得到不连续的固溶体即为低共熔物[13]。图6显示了分别以MeNQ和NQ为基础分子模拟得到的能量最低的配位模式。可见在MeNQ分子周围最多可配位6个NQ分子,而NQ分子周围则最多可配位5个MeNQ分子。
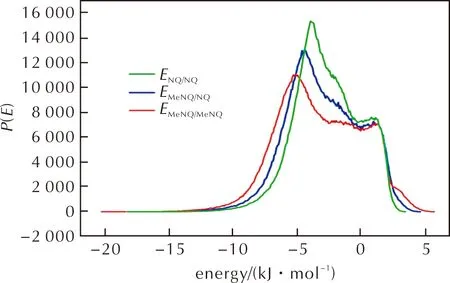
图5 MeNQ和NQ体系的作用能分布曲线Fig.5 The interaction energy distribution of MeNQ and NQ
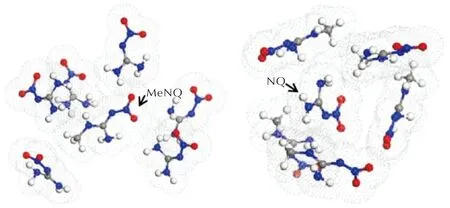
图6 基础分子与检测分子能量最低的配位模式(网点部分为分子的van der Waals表面)Fig.6 The lowest energy coordination mode between the basic molecule and the detection molecule (shaded area represents the van del Waals surface)


图7 MeNQ/NQ体系的ΔEmixAB与温度T的变化曲线Fig.7 Temperature dependent curves of ΔEmix and AB for MeNQ/NQ system
通过上述结构和作用能分析可知,MeNQ和NQ为非同晶材料,且分子间存在尺寸和形貌上的失配。第一性原理和Monte Carlo模拟均说明,MeNQ与NQ二元体系中分子间的附着力弱、内聚力强。根据Cherukuvada[12-14]和Mohamed[46]等的研究结果,可预测MeNQ和NQ的共结晶产物应为低共熔物。实验上尚无MeNQ/NQ低共熔物制备与表征的详细报道。目前李华荣[41]报道了MeNQ/NQ二元相图的绘制。分别采用熔融法和研磨法,基于DSC测试数据和van′t Hoff方程,绘制得到的相图均呈“V”字形,一种典型的低共熔物相图特征。通过绘制的相图可直接得到低共熔点的组成和温度,MeNQ与NQ摩尔比为7∶3,温度约130℃。该实验研究也说明MeNQ和NQ共结晶产物为低共熔物。
3 结 论
(1)基于静电势互补和氢键供体-受体匹配原则,构建了MeNQ与NQ的二聚体模型,在M062X-D3/6-311+G(d, p)水平上对其进行结构优化,得到能量最低的二聚体结构。采用精度更高的计算方法(MP2/may-cc-pvtz和PWPB95/ma-def2-QZVPP,得到最稳定结构的分子间相互作用能。结果表明,二聚体MeNQ-MeNQ的分子间作用力大于MeNQ-NQ及NQ-NQ而占据支配地位。在sSAPT0/jun-cc-pVDZ水平上的能量分解结果表明,3种二聚体中分子间的吸引项主要为静电作用力,其次为诱导和色散力,交换能为排斥项。吸引力和排斥力的协同作用导致MeNQ分子间的内聚力最强。
(2)MeNQ属于单斜晶系,而NQ属于正交晶系,二者为非同晶材料。在M062X-D3/6-311+G(d, p)水平上的分子结构优化结果表明,甲基的引入破坏了NQ分子的平面结构,分子体积也从NQ的68.418cm3/mol增加到MeNQ的81.902cm3/mol,分子间存在形貌和尺寸上的失配。
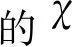
猜你喜欢
航空材料学报(2023年6期)2023-12-18 05:23:50
小学生学习指导(小军迷联盟)(2023年3期)2023-03-27 09:22:44
广州化工(2022年19期)2022-11-09 11:30:46
广州化工(2022年18期)2022-10-22 10:27:00
中国音乐学(2022年1期)2022-05-05 06:48:46
建材发展导向(2020年15期)2020-11-26 12:55:22
工业催化(2020年5期)2020-06-23 01:59:12
硅酸盐通报(2020年1期)2020-02-25 10:01:30
四川水泥(2019年9期)2019-02-16 20:12:56
物理学报(2018年10期)2018-06-14 06:31:32
