
Cre-LoxP条件性基因敲除的实际应用策略
2022-10-26 03:49孔梓宇
中国生物化学与分子生物学报 2022年9期
孔梓宇, 柳 毅, 汪 晖,2)*
(1)武汉大学基础医学院药理学系, 武汉 430071; 2)湖北省疾病发展与防治重点实验室, 武汉 430071)
基于Cre-loxP系统的基因敲除具有操作简单、传代相对稳定等优点,但在实际应用Cre-loxP系统时,发现其存在缺陷,例如Cre重组酶的毒性、Cre重组酶的“异位”表达等。为了解决这些问题,人们改进设计方案,并不断优化Cre-loxP系统,使其能够更好地适用于各种条件下的敲除目的。本文从Cre序列的构建、flox序列的构建以及条件性敲除(conditional knockout, CKO)动物的繁育等方面综述了这些实际应用方案,并总结了一些能够更好地控制敲除时间空间特异性的衍生Cre-loxP系统,包括不依赖他莫西芬的可诱导Cre重组酶、高活性Cre重组酶以及光信号介导的Cre重组酶等[1-3]。最后,本文展望了未来Cre-loxP系统的发展前景,对Cre-loxP系统的进一步成熟、可控性的提升提供新思路。
1 Cre-loxP系统
Cre-loxP系统起源于P1噬菌体。Cre重组酶是一类由343个氨基酸构成的重组酶,于1981年在P1噬菌体中被发现,属于λ Int酶超基因家族,能够特异性识别loxP位点,并发挥重组酶活性或限制酶活性[4]。当目标基因序列的两侧嵌入同向的loxP位点时,Cre重组酶会特异性识别loxP位点并使其成环断开,实质上造成了loxP位点之间的基因被敲除[5]。loxP位点同样来源于P1噬菌体,长度为34 bp,包括了2个回文序列和中间8 bp的核心序列。回文序列是Cre重组酶的特异识别位点,而核心序列则决定了loxP位点的方向性[6]。
Cre重组酶特异性识别回文序列并结合形成二聚体,此后2条染色体上同时形成二聚体并再次结合形成四聚体。此时2个loxP位点间的DNA序列会被Cre重组酶切断。紧接着,DNA连接酶会快速高效地将这些序列连接起来。根据loxP位点的位置和方向[7],重组结果可分为以下3种情况:如果2个loxP位点位于同一条DNA链上且方向相同,Cre重组酶介导loxP间的序列切除;如果2个loxP位点位于同一条DNA链上且方向相反,Cre重组酶介导loxP间的序列反转;如果2个loxP位点位于不同的DNA链或染色体上,Cre重组酶介导DNA链发生交换或染色体易位[8]。
自80年代起,基因敲除逐渐发展,世界已涌现出各种基因编辑的技术。例如,锌指核酸酶(zinc-finger nucleases,ZFN)技术、转录激活样效应因子核酸酶(transcription activator-like effector nucleases,TALEN)技术等。但这些技术操作工序繁琐,且不具有组织特异性。而全身性敲除极有可能导致胚胎的致死率过高,同时难以研究某一器官或组织中某一基因的功能。因此,选用基于Cre-loxP系统的基因敲除,可以通过组织特异性启动子与该系统的联用,实现组织特异性基因敲除,这大大拓宽了基因敲除的空间可塑性与应用前景。
2 Cre-loxP的实际应用
虽然Cre-loxP系统的横空出世实现了基因编辑技术的又一次飞跃,然而在实际应用Cre-loxP系统时,仍然发现其存在潜在的缺陷[9]。以小鼠为例,规避这些问题需要从Cre工具鼠的构建、flox工具鼠的构建以及条件性敲除繁育等方面逐一考虑。
2.1 序列的插入策略
在进行工具鼠构建时,CRISPR/Cas9系统常被应用于靶序列插入环节。载体构建完成后,经尾静脉注射或胚胎干细胞注射技术可以构建Cre工具鼠或flox工具鼠。在实际应用过程中,靶序列的长度、位置、插入方式等特性都需要根据要求进行个性化设计,以避免无效插入。
2.1.1 CRISPR系统在插入序列中的应用 目前,常用CRISPR/Cas9系统将所需的序列插入表达载体中。CRISPR/Cas9系统起源于细菌,是细菌在长期进化中逐渐形成的免疫防御系统。CRISPR/Cas9系统包含一种被称为sgRNA的单向导RNA,由靶标特异的crRNA和通用的tracrRNA组成。tracrRNA招募的Cas9核酸酶可以识别靶基因PAM序列,并在其上游3~4bp位置进行剪切而形成双链断裂。断裂的DNA若采用非同源末端连接的方式进行修复,则可实现基因敲除;反之,若采用同源重组的方式进行修复,则可实现基因敲入[10]。人们对受精卵进行特殊处理可以使细胞倾向于采用同源重组的方式进行修复,从而实现Cre编码基因的插入。相比于其他的基因编辑技术,基于CRISPR/Cas9系统的序列插入具有较大的优势,这主要因为CRISPR/Cas9技术问世前的定向打靶都依赖于DNA序列特异性结合蛋白质模块的合成,这一步骤非常繁琐费时;而CRISPR/Cas9技术使用一段序列特异性向导RNA分子引导核酸内切酶到靶点处,显著缩减了前期设计成本。同时,基于CRISPR/Cas9系统的基因编辑是较为可靠的,脱靶概率较低[11]。因此,CRISPR/Cas9系统被认为是十分便捷且高效的动物模型构建工具[11],对小鼠基因组工程的实施产生了重大影响[12,13]。
2.1.2 Cre的插入策略 Cre编码序列的插入位置一般有2种:原位插入和安全位点插入。原位插入是指根据敲除目的合理选择上游的启动子序列,然后将Cre编码序列插入至靶基因附近的插入方法。在实际操作中,若需进行全身敲除,则可在Cre编码序列前连接管家基因以保证全身性的Cre酶表达;有时需要针对某基因进行组织特异性敲除,此时要在Cre编码序列前连接组织特异性表达基因,以保证Cre序列与靶基因共用启动子区域。第2种插入方法为安全位点插入。早在20 世纪 90 年代人们就发现,ROSAβgeo26的随机转基因小鼠品系在所有组织中都能检测到高水平的β半乳糖苷酶表达。由于Rosa26位点能保证外源基因的正常稳定表达,因此,人们称其为安全位点[14]。在此,本文给出了一种典型的Rosa26基因敲入的载体设计图(Fig.1)。此后,又陆续发现了许多安全位点,例如H11、CCR5、AAVS1等位点[15, 16]。其中,H11位点位于小鼠第11号染色体的Eif4enif1与Drg1基因之间[17],且这2个基因的侧翼序列能够使整合在此的外源基因受指定启动子的驱动而稳定表达。并且H11位点的纯合插入小鼠也能实现正常的发育与繁殖,这使得H11成为了继Rosa26之后最受欢迎的安全位点。
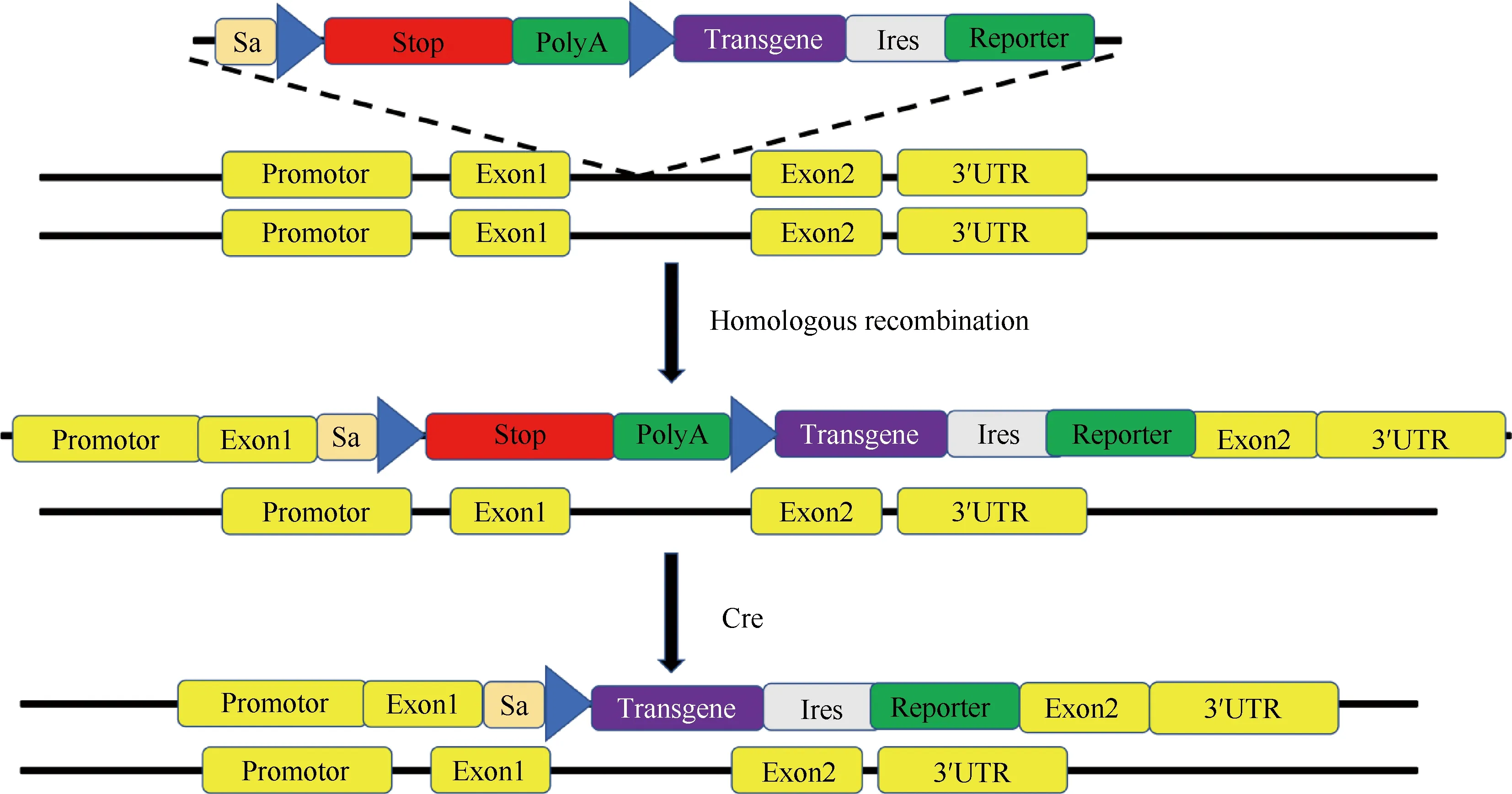
Fig.1 A typical ROSA26 knock-in vector design The combined exogenous sequence is integrated into the ROSA26 safety site through homologous recombination. When the recombinant enzyme Cre comes into play, the stop sequence will be cut off, thus driving the expression of the target sequence
安全位点除了可驱动靶基因的稳定表达以外,还能用来规避cre重组酶与loxP位点位于同一染色体上的问题。当Cre编码序列与loxP序列位于同一染色体的时候,除非发生预期外的重组,否则后续的繁育无法正常获得符合条件的动物(Fig.2)。此时,采用在安全位点进行基因敲入的方式,可将Cre序列转移至非连锁基因座上,从而避免了上述情况。因此,安全位点的发现有效解决了Cre与loxP连锁遗传的问题。
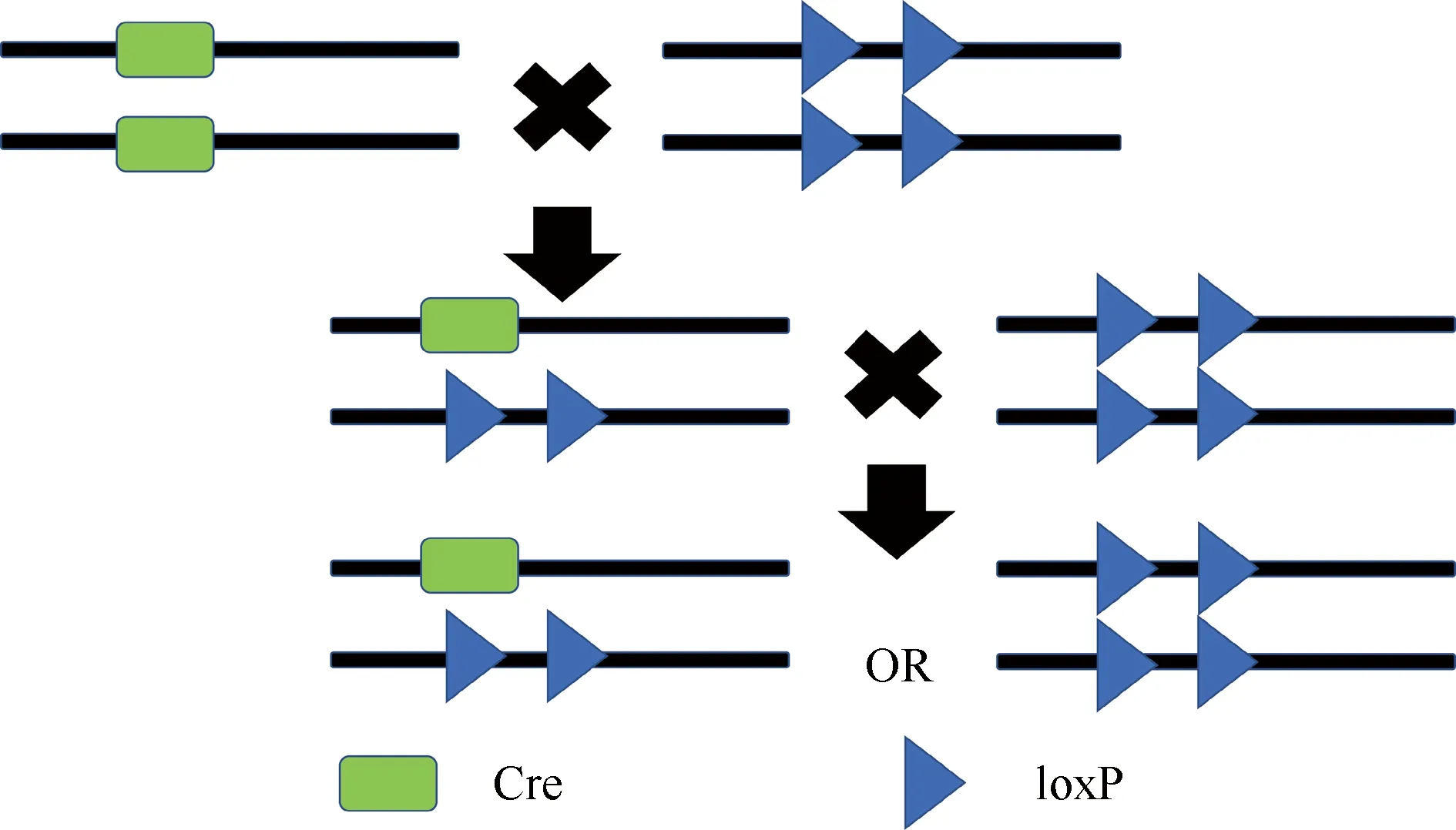
Fig.2 Genetic representation when Cre and LoxP on the same chromosome When the Cre encoding sequence and loxP sequence are located on the same chromosome, we can never obtain mice containing both Cre and allelic loxP sequences unless an unexpected recombination occurs
在真核生物中,绝大多数基因都存在多个外显子与内含子,如何选择合适的位置插入Cre编码序列取决于后续研究的需要。通常来说,Cre重组酶插入在基因第1个外显子前可以保证Cre重组酶的表达量,但可能会影响下游基因的正常表达;而将Cre编码序列插入在基因的最后1个外显子下游,则会起到对原基因的保护作用,却又可能导致Cre重组酶的表达量降低。由于现在人类尚未研究清楚每一个具体的插入位点会带来什么样的分子生物学影响,因此在无法查询到相关信息的时候,一般根据自身的需要以及前人的经验来选择Cre重组酶的插入位点。此外,Cre编码序列尽量不要跨过内含子区域,以防止对序列的表达产生不可预知的影响。
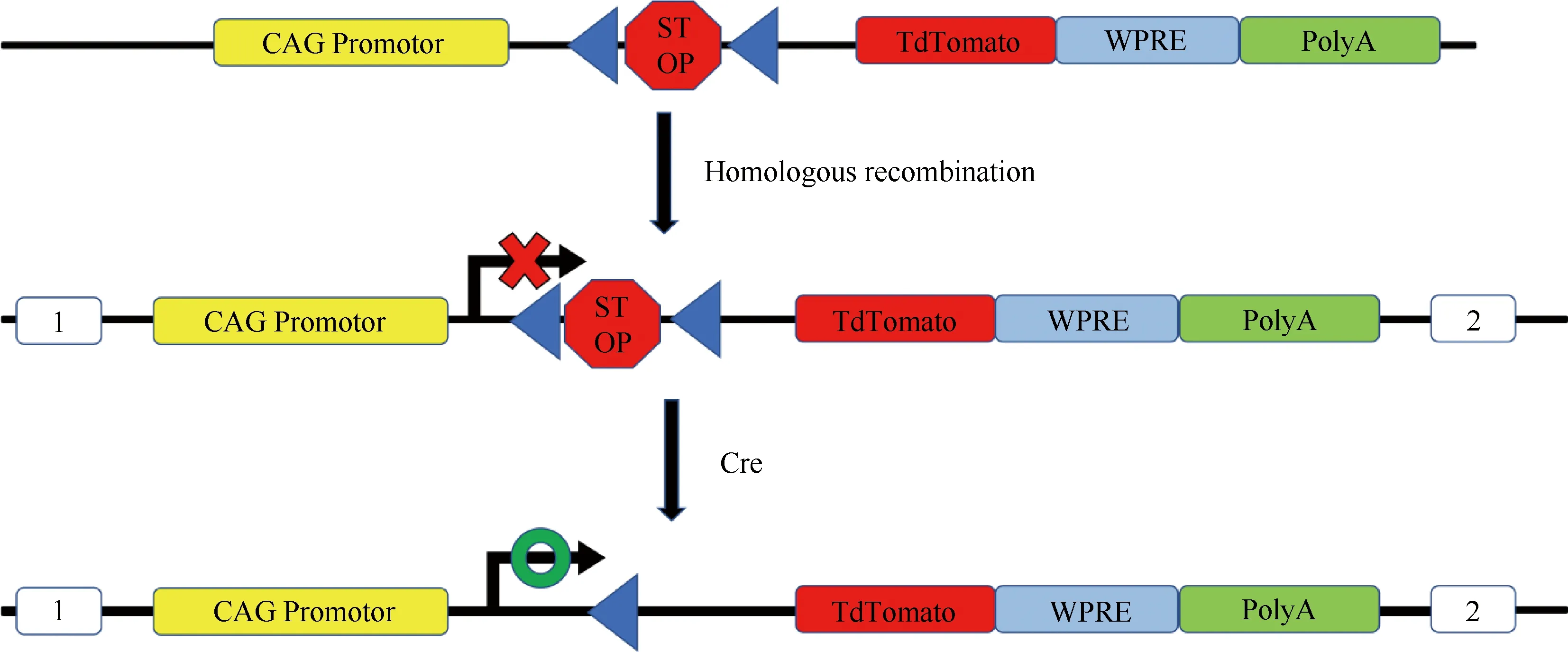
Fig.3 A schematic diagram of a typical TdTomato sequence insert scheme The STOP sequence expressing Cre enzyme was cleaved and the red fluorescent protein was expressed. By observing the red fluorescence of animals, it can be determined whether there is leakage expression
2.1.3 loxP的插入策略 基于Cre-loxP系统的基因敲除需要在感兴趣的基因上下游插入2个同向的loxP序列。然而,实际操作过程中可能会遇到基因敲除致死的情况发生,此时应适当减小loxP位点间的距离。例如,对NR3C1进行组织特异性敲除的时候,一般仅在单一外显子的两侧插入loxP位点[18]。在这种情况下,外显子的选择可根据自身研究的需要进行调整。当loxP位点下游有多个外显子时,应考虑Cre酶介导的同源重组是否会带来移码突变,此时为了降低致死率,应尽可能避免移码突变的发生。
2.2 序列的验证试验
在设计Cre表达载体的时候,需要设计1个或多个表达标签蛋白质的序列来进行后续验证。常用的标签蛋白质有绿色荧光蛋白(green fluorescent protein, GFP)等。这些蛋白质的编码序列需要与Cre序列嵌合插入并共同表达,才能满足后续验证的需要。在设计标签蛋白的插入位置时,若插入的蛋白质是含有信号肽的蛋白质,则尽量在C-端插入而避开N-端。因为N-端的标签会跟随信号肽一同被剪切,从而丧失验证的功能。
Cre重组酶通常靶向成年小鼠中少量细胞中表达的基因。然而,在生殖系或发育早期可能存在Cre重组酶的短暂表达[19]。这种非正常现象被称为“异位”,其发生概率较高,并且少量Cre重组酶作用于loxP位点便可以激发。当Cre重组酶存在于男性生殖系中时,可能会有少量的Cre重组酶通过精子传递到发生重组的卵子中。当Cre重组酶在卵母细胞中表达时,即使在受精后也能发生重组[20]。通过与荧光标记flox动物交配可以对Cre重组酶的表达进行验证。相比较而言,这种验证能更精确地获取Cre酶的空间特异性表达情况[21]。例如,在TdTomato荧光蛋白上游添加loxP-STOP-loxP序列,并利用CAG启动子介导转录,可将整个片段插入在Rosa26位点上(Fig.3)。在Cre重组酶表达前,STOP序列的存在阻止了红色荧光蛋白质的表达,而与含有组织特异性Cre酶进行交配繁育后,其子代同时具有Cre重组酶与loxP位点,表达Cre酶的细胞内STOP序列被剪切,红色荧光蛋白质得以表达。而通过观察F1代动物的红色荧光,可以确定所构建的Cre工具动物是否存在泄漏表达的情况[22]。
在设计针对flox动物的验证实验时,常用的是在mRNA水平上进行验证。针对flox工具鼠,研究通常在靶标序列两侧的loxP位点外设计2个引物[23]。在验证时,选取小鼠尾部部分组织进行DNA提取,随后逆转录相应的携带或不携带loxP标签的靶序列,再进行琼脂糖凝胶电泳(Fig.4)。由于所携带的loxP标签的序列长度大于未携带的loxP标签序列,因此,在凝胶中泳动距离较短的条带为flox序列,反之为阴性序列。
在针对CKO敲除鼠进行验证实验时,同样可以在loxP位点外侧设计2个引物。但实际操作过程中,敲除的片段经常过大而导致其无法被正常扩增,此时也可以选择在loxP位点内部再额外设计一个引物[24, 25](Fig.5)。
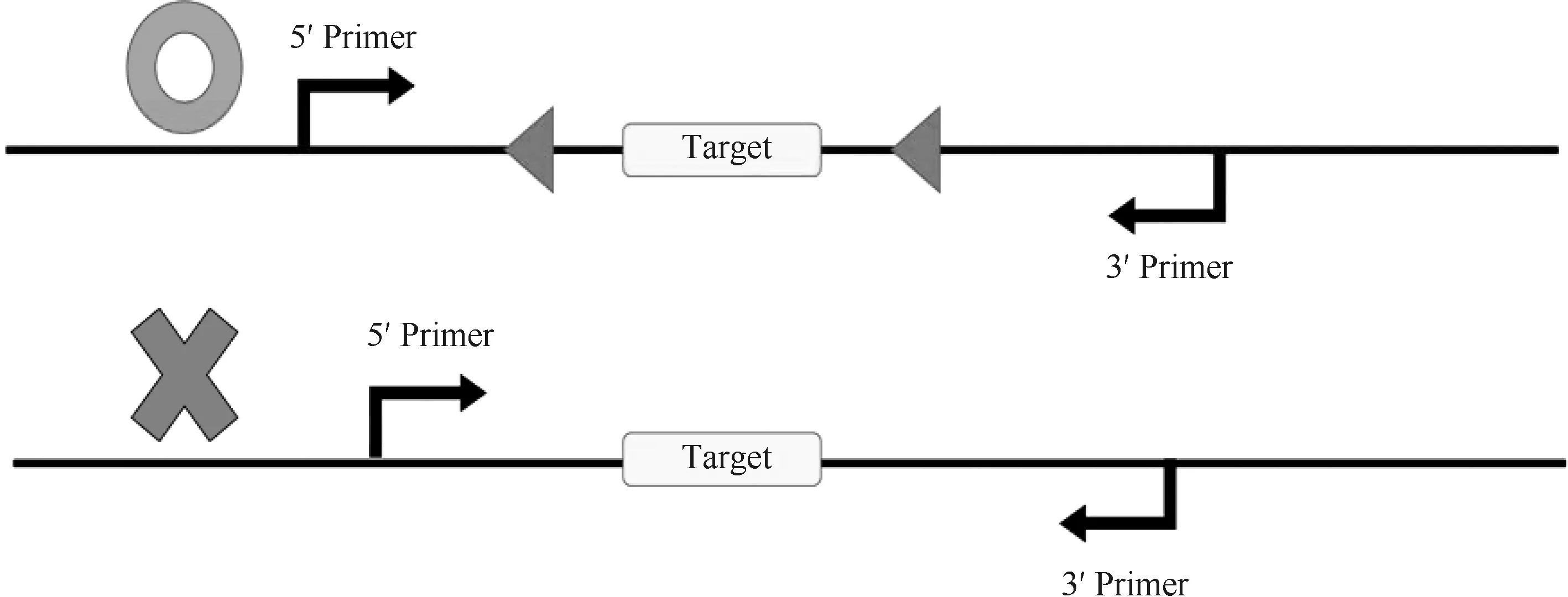
Fig.4 Verification design of flox tool mouse Two primers were designed outside the loxP sites on both sides of the target sequence, and the genotypes of the animal could be determined according to the size of the bands in agar-gel electrophoresis. Two strips mean homozygous; A short band and a long band mean heterozygous; Two bands mean negative
除了上述基于mRNA水平上的验证,还可以进行蛋白质水平上的验证。常用方法为设计相应的抗体,进行Western印迹、免疫组化以及免疫荧光等实验。
2.3 条件性敲除繁育
在获得符合条件的Cre工具鼠与flox工具鼠后,需要将两种工具鼠进行交配繁育,以获得实验动物。常规的交配方式,一般采用flox纯合小鼠与Cre杂合小鼠进行交配,其后代会产生半数flox(+/-)cre(+/-)与半数flox(+/-)cre(-/-)小鼠[26]。一般在利用Cre-loxP系统进行基因敲除时,不需要Cre处于纯合状态。这是由于Cre的杂合状态已经足以表达一定量的Cre重组酶,并且Cre的纯合可能会导致其等基因座的序列被彻底破坏,从而产生不可预知的结果[27]。此外,研究表明,Cre重组酶在发育精子中的表达会导致雄性不育,这可能是Cre介导的基因组重排所致[28]。
在利用Cre-loxP系统构建组织特异性或者细胞特异性基因敲除的时候,需要考虑到Cre泄漏表达的问题[29]。例如,研究显示,Tg(Thy1-cre)1Vln(Thy1-cre)原本被认为仅在大脑皮质与海马神经元内表达[30]。但后来发现,心肌、血管内皮、肺泡和细支气管细胞、皮肤以及睾丸等器官中均有cre表达[8]。值得注意的是,一些生殖细胞特异性表达的Cre或一些在生殖细胞中“异位”表达的Cre,会导致其子代的靶序列全身性敲除,破坏了原有的实验模型[31]。此时,可以采用单性别繁育,即仅利用某单一性别小鼠作为Cre工具鼠[20]。
3 Cre-loxP系统在条件性基因敲除中的优化应用
Cre-loxP重组系统目前已广泛应用于生物医学研究。然而,最初无法对这种重组进行高精度时空控制[3]。为此,研究尝试开发新型Cre-loxP系统以实现对基因编辑的时空特异性。
3.1 时间可诱导型Cre
重组酶Cre的表达可以在空间上由组织特异性启动子区域调控,而在时间上会由外源诱导物调控。他莫昔芬系统和四环素系统是目前常用的两套在时间上控制Cre重组酶表达的系统。此外,光等物理因素也能作为诱导信号对Cre的表达状态进行控制。
3.1.1 配体诱导型Cre 最初的研究发现,将Cre与人类雌激素受体(estrogen receptor, ER)的配体结合区结合形成的复合体,只有在雌激素诱导后才会与锚定蛋白质HSP90脱离,从而入核发挥作用。为了避免内源雌激素的干扰,在雌激素受体ER的配体结合区(ligand binding domain, LBD)做一个点突变(G521R)就可以使Cre-ER只响应外源的人工合成雌激素的诱导,命名为Cre-ERT。Cre-ERT的出现使研究胚胎致死基因在发育后期的功能得以实现,并且可以利用这种小鼠系进行谱系示踪[32]。而另一种LBD 突变体融合蛋白质被证明,其对4-OHT具有远高于Cre-ERT的敏感性,这种突变体是Cre-ERT2,它带有人ER LBD中的3个点突变:C400V/M453A/L544A。目前,新研发的Cre-ERT2比Cre-ERT效率更高[33]。但即使是优化后的Cre-ERT2系统,也不乏其泄露表达的报道[34, 35]。事实上,Cre-ERT2的泄漏表达是随机发生的,但其发生概率与Cre-ERT2分子丰度、染色质状态甚至loxP的物理定位都有关系[32]。因此,在设计谱系追踪和脉搏追踪实验时,应该考虑到与他莫西芬无关的Cre-ERT2活性的影响。为了降低这种背景影响,可以使用重组阈值相对较高的报告基因,例如mTmG或R26R-EYFP等[36]。
3.1.2 启动子激活型Cre 与配体诱导型Cre不同,启动子激活型Cre通过驱动Cre重组酶的转录启动来操纵基因敲除。例如,四环素系统是基于四环素操纵子的系统。由于四环素阻遏蛋白质(tetracycline repressor protein, tetR) 与四环素操纵子序列(tetracycline operator DNA sequence, tetO)的自然结合状态可以阻止四环素抗性基因表达,Tet的存在能终止tetR和tetO的结合状态,从而解除四环素操纵子的阻遏作用[37]。后来,根据此原理开发了Tet-on系统与Tet-off系统等基因表达调控系统。在Tet-on系统中,不给予Tet时目的基因处于表达状态,而在Tet-off系统中,给予Tet时目的基因处于表达状态[38-40]。与Cre-ERT类似的是,此类时间诱导型Cre-loxP系统能规避大量Cre酶带来的细胞毒性,但对于增殖速度极快的细胞仍然具有较强毒性。因此,在解释快速增殖的细胞相关研究时,要谨慎对待Cre-loxP系统的相关实验数据[41]。
同时,干扰素系统也是启动子激活型Cre的重要组成部分。通过干扰素,例如pIpC等可以诱导Mx1基因启动子的激活,从而驱动Cre重组酶的表达。然而,干扰素系统驱动的Cre模块容易自发重组,并且pIpC本身便会引起不良的副作用。因此,在血液学研究等领域中,对Mx1-Cre系统驱动的基因敲除需要进行稳态控制。例如,开发新诱导物、移植诱导等方法[42]。
3.1.3 光诱导型Cre 与化学诱导剂可能具有细胞毒性、泄露表达的缺陷相比,光是一种很好的时空调控基因表达的诱导剂。然而,并非所有波段的光都适合作为诱导剂。蓝光的光诱导型Cre酶,例如CRY2-Cre系统中,2个Cre片段融合到对蓝光敏感的植物光受体隐花色素2 (cryptochrome 2, CRY2)或其结合域CIB1上。其中,一种组分与CRY2融合,另一种组分与CIB1融合,被蓝光照射会引起异二聚化[3]。但这种蓝光进行诱导时,由于其组织穿透性不强,因此,需要较长时间进行诱导。考虑到蓝光等短波长光源对细胞的光毒性,此类光诱导型Cre酶的引用因此受到了局限;同样,常规的基于紫外光诱导的光诱导型Cre存在较强的光毒性,可能给细胞带来影响[43]。
研究表明,较长的波长光源一般具有更好的诱导能力。例如,远红外光源相比于常规光源的组织穿透性更强[1]。因此,利用长波长光作为诱导光源能显著缩短诱导时间,可能会大幅度降低对细胞的光毒性。在一种远红光诱导的Cre-loxP系统中,Cre重组酶分为2个片段,N-端Cre片段融合到Coh2结构域,而C-端Cre片段融合到DocS区域。该融合蛋白质的诱导表达由frl反应启动子PFRLx驱动。在FRL光照下,2个片段结合在一起时,可以根据Coh2和DocS域的强亲和力,重新构建Cre重组酶的活性[44]。该系统对小鼠无明显的细胞毒性作用,重组效率也有了大幅度提升。
总之,将Cre-loxP系统与相关其他分子生物学工具联用可以获得其时间空间可控性,从而满足各种条件的敲除需要。
3.2 活性改造型Cre
对Cre元件的改造显著提升了Cre重组酶的活性。例如,通过在Cre元件上引入真核细胞核定位序列NLS,此时的Cre重组酶能在低表达丰度下实现重组。这对于一些低丰度的启动子十分重要。此外,有研究提及了sCreER系统,该系统具备转换成其他活性形式Cre的能力,从而显著提升DNA重组效率[45]。在R26-LZLT、R26-GFP或Kdr等位基因等重组相对惰性的基因位点上,sCreER都能有效介导基因重组。凭借这一独特的优势,sCreER将进一步推动生物医学研究。例如,通过R26-tdTomato报告基因发现,Npr3-sCreER和Col1a2-sCreER分别有效靶向大多数心内膜细胞和成纤维细胞[44]。同时,sCreER经过一次他莫西芬诱导后,可以转换成Cre,这将显著降低他莫昔芬本身带来的毒性,从而避免动物表型受其干扰。
4 问题与展望
总之,基于Cre-loxP系统的基因敲除是基因编辑发展史上重要的里程碑。它显著缩减了基因敲除的繁杂步骤,也更加适用于对特定组织或细胞种类进行特异性基因敲除,明显拓宽了基因敲除的应用范畴。同时,这种基因敲除技术在实际操作方面存在大量需要考虑的问题。例如,Cre重组酶的泄漏表达、序列插入时的脱靶现象、非预料的重组与敲除效率不稳定等。人们通过不断优化Cre-loxP系统本身及其具体设计策略,不仅改善了上述情况,还实现了Cre-loxP系统的时间空间可控性。可以预见的是,未来基于Cre-loxP系统的基因敲除会倾向于优化Cre重组酶的特异性表达、优化Cre重组酶的重组效率、提高重组精准性与规避Cre重组酶的毒性。此外,还会尝试将Cre-loxP系统的应用范围扩大,以适应更多的研究需要。例如,有研究指出,Cre-loxP系统与类似的Dre-rox系统联用,会更精确地调控重组酶的表达,提高细胞谱系示踪的精确性[46]。总而言之,在不久的将来,位点特异性重组系统将广泛应用于对基因操作的高分辨率时空控制有极大需求的生命科学研究领域[47]。
猜你喜欢
上海金属(2021年6期)2021-12-02
数学物理学报(2021年4期)2021-08-30
昆明医科大学学报(2021年3期)2021-07-22
中国生殖健康(2020年2期)2021-01-18
新世纪智能(数学备考)(2020年10期)2021-01-04
生物学通报(2019年3期)2019-02-17
电脑知识与技术(2018年19期)2018-11-01
中成药(2017年12期)2018-01-19
中国交通信息化(2017年8期)2017-06-06
中国医疗保险(2017年5期)2017-05-17
- 中国生物化学与分子生物学报的其它文章
- PM2.5通过激活NLRP3/Caspse-1通路诱导大鼠子宫炎症反应
- “金课”背景下生物化学课程教学的创新与实践
- EHD2 Affects the Proliferation of Esophageal Squamous Cell Carcinoma by Regulating the Cyclin D1-CDK4-pRb Signaling Axis
- Therapeutic Effect of Mesenchymal Stem Cells Overexpressing Interleukin-10 on Inflammatory Bowel Disease
- miR-31改善2型糖尿病小鼠的肝损伤
- 干扰NSUN2通过调控细胞周期蛋白表达抑制黑素瘤细胞增殖