
DNAH5基因突变致儿童原发性纤毛运动障碍一例报告及文献复习
2022-10-13 12:58包贝贝宋桂华彭明浩于素平周鸿雲
郑州大学学报(医学版) 2022年5期
包贝贝,宋桂华,彭明浩,刘 洁,于素平,张 岩,周鸿雲
1)河南中医药大学儿科医学院 郑州 450000 2)河南中医药大学第一附属医院儿科 郑州 450000
原发性纤毛运动障碍(primary ciliary dyskinesia,PCD)是一种罕见的常染色体隐性遗传病[1],因活动纤毛结构异常和(或)功能障碍导致黏液纤毛清除和黏液滞留功能不佳[2]。新生儿呼吸窘迫、慢性化脓性肺部疾病、慢性浆液性中耳炎和慢性鼻窦炎是PCD常见的呼吸道表现[3]。大约50%的PCD患者有内脏转位,临床上也常见不孕症[4]。Guo等[5]发现50例确诊为PCD的中国患儿中约70.0%有内脏转位,明显高于欧洲和美国患儿。目前已知与PCD相关的基因已超过40个,最常见的导致PCD的基因是编码外动力素臂(outer dynein arms,ODA)蛋白的DNAH5和DNAH11[6-7]。本文回顾分析1例DNAH5基因突变致PCD患儿的临床资料,并结合文献进行复习,以提升对该病的认识。
1 临床资料
1.1 一般资料患儿,女,12岁6个月,因“反复咳嗽、咳痰2 a余,加重半个月,发热1 d”于2021年5月7日收入河南中医药大学第一附属医院儿科。患儿于2 a前出现反复咳嗽,咳痰;半个月前出现咳嗽、咳痰加重,伴鼻塞、流黄涕、胸闷、喘息,自行口服头孢克肟颗粒治疗3 d,布地奈德混悬液+特布他林溶液雾化吸入1周,症状有所缓解;3 d前于河南中医药大学第一附属医院查肺部CT提示:双肺多发支气管扩张合并感染;双侧胸膜增厚粘连。予中药颗粒(具体不详)口服,治疗效果欠佳,1 d前患儿发热,热峰40 ℃,自行口服头孢克肟颗粒、连花清瘟胶囊治疗,随后体温恢复至正常,精神差、咳嗽、咳痰、胸闷、喘息,遂收入我院治疗。
个人史:患儿为足月顺产,出生体重3.75 kg,否认窒息抢救病史,出生后配方奶粉喂养,适龄添加辅食,生长发育同正常同龄儿。
家族史:父母体健,非近亲婚配;妹妹11岁6个月,既往鼻窦炎史、反复“支气管扩张”,当地医院治疗好转,既往检查示内脏转位(家属口述,未见检查单);弟弟5岁8个月,出生时曾入住新生儿病房(具体治疗不详),自2020年始咳痰多,偶尔口服中药治疗,效果一般。
既往史:患儿出生后有黄疸史、肺炎史,于当地医院住院治疗1周(具体治疗不详)。2021年1月28日在当地医院诊断为“肾型过敏性紫癜”,分别于当地医院及我院儿科住院治疗,病情好转后出院。
入院查体:体温36.8 ℃,呼吸20次/min,心率92次/min,血压100/64 mmHg(1 mmHg=0.133 kPa),体重45 kg。咽腔充血,双侧扁桃体无肿大,表面未见脓性分泌物;肺下界正常,听诊双肺呼吸音粗,可闻及少量湿性啰音及痰鸣音;心腹未查及明显异常;神经系统未查及异常体征;双下肢散在点状色素沉着斑,呈褐色,无抓痕,无破溃、渗液等。
影像学检查。肺部CT(图1A)示:①双肺多发支气管扩张合并感染。②双侧胸膜增厚粘连。常规心电图、心脏彩超均未见异常。腹部彩超示:脾稍大,余未见明显异常。电子支气管镜检查(图1B)示:①支气管扩张(左肺舌叶各段和右肺中叶各段管腔扩张,管壁呈鱼骨刺样改变)。②支气管内膜炎(左肺舌叶各段、右肺中叶各段及下叶内基底段管腔内条絮状分泌物栓塞,予灌洗治疗)。
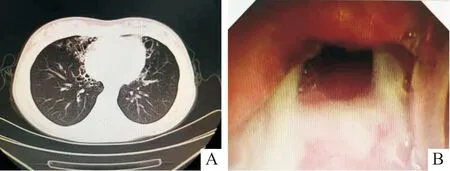
图1 患儿肺部CT(A)和电子支气管镜表现(B)
住院期间该患儿诊断为:①肺炎。②支气管扩张。③全身炎症反应综合征。④肾型过敏性紫癜。
1.2 DNAH5基因检测与家系情况
1.2.1DNAH5基因检测 住院期间经患儿父母知情同意,抽取患儿、其父母和弟弟、妹妹外周血2 mL行临床全外显子测序(上海韦翰斯生物医药科技有限公司),2021年6月15日基因检测结果(图2)发现患儿DNAH5基因(NM_ 001369.3)2个杂合突变:63号外显子c.10616G>A(p.R3539H)和61号外显子c.10363G>T(p.Q3455*)。前者为DNAH5基因的第3539位密码子由编码精氨酸变成编码组氨酸,为已知致病突变[8-11];后者为DNAH5基因的第3455位密码子由编码谷氨酰胺变成终止密码子,可能导致基因功能缺失,为新发突变,未见文献报道,根据ACMG临床实践指南判定为致病[12-13]。

A、B、C:分别为患儿及其弟弟、妹妹DNAH5基因63号外显子c.10616G>A及61号外显子c.10363G>T杂合突变;D:患儿父亲DNAH5基因61号外显子c.10363G>T杂合突变;E:患儿母亲DNAH5基因63号外显子c.10616G>A杂合突变
1.2.2家系情况 家系图见图3。患儿及其弟弟、妹妹的这两个突变分别遗传自母亲和父亲,其父亲只携带后1种突变,其母亲只携带前1种突变,患儿及其弟弟、妹妹为这两个突变构成杂合突变。

P为患儿;N:基因检测正常;M1:DNAH5基因63号外显子c.10616G>A(p.R3539H);M2:DNAH5基因61号外显子c.10363G>T(p.Q3455*)
1.3 治疗经过7 d抗感染(头孢曲松钠静脉滴注)、雾化(先后给予布地奈德混悬液、硫酸特布他林雾化液、复方异丙托溴铵溶液、吸入用乙酰半胱氨酸溶液)、对症化痰(氨溴索静脉滴注)、中药等治疗,患儿精神状态好转,咳嗽、咳痰症状明显减轻,肺内啰音基本消失。白细胞计数8.0×109个/L,中性粒细胞百分比79.0%,淋巴细胞百分比14.9%,CRP 4.3 mg/L。肺泡灌洗液细菌(嗜血杆菌、抗酸杆菌等)、真菌涂片,肺泡灌洗液嗜血杆菌、抗酸杆菌培养,儿童血嗜血杆菌、抗酸杆菌培养均无异常。肺功能测定:肺通气功能在正常范围,运动后1 s用力呼气容积(FEV1)下降率为12.9%(在正常范围内)[14]。呼出气一氧化氮(NO)测定结果:FeNO 8 mm3/m3(在正常范围内),CaNO 6.5 mm3/m3(>5 mm3/m3),反映小气道炎症[15]。患儿于2021年5月15日出院,嘱患儿及家属定期至门诊复诊。
2 讨论
人体中纤毛由200余种蛋白质构成,分布广泛且作用重要,可分为运动纤毛和非运动纤毛两种类型,前者广泛分布于呼吸系统、输卵管和脑室等部位,驱动机体运动中上皮细胞上的液体运输,如胚胎器官的左右模式形成、呼吸道黏液和污垢的清除以及精子和卵子的正常运动[16]。运动纤毛的横切面在电镜下呈现“9+2”的结构形态,其正常运动由纤毛的内、外动力臂共同通过ATP酶活性起作用[17]。活动纤毛的结构异常或功能受损可导致多种纤毛疾病,如PCD、多囊肾病、脑积水、心脏缺陷等[16]。有研究[18]对78例被诊断为PCD患者的纤毛进行透射电子显微镜分析发现纤毛最明显的缺陷主要是内、外动力蛋白臂和中央装置的缺陷。
PCD是一种遗传异质性疾病,多为孟德尔常染色体隐性遗传,但也有报道常染色体显性遗传及X染色体连锁遗传可导致本病[19]。新生儿主要表现为不明原因的呼吸窘迫,后逐渐出现婴儿期持续湿咳、慢性鼻窦炎、分泌性中耳炎和支气管扩张,部分患者会出现内脏转位、不孕症或不育症[20]。PCD 包括纤毛不动综合征、Kartagener综合征(Kartagener syndrom,KS)、纤毛运动不良和原发性纤毛定向障碍等几个类型。KS以支气管扩张、内脏转位和慢性鼻窦炎三联征为特征[21],该患儿临床检查未见内脏转位,据父亲口述患儿妹妹既往检查示内脏转位,结合基因检测结果可考虑KS。
DNAH5被认为是PCD中突变频率最高的基因[22]。DNAH5是在纤毛中发现的微管相关蛋白复合物的一部分,显著高表达于支气管与肺组织中,编码一种与纤毛外动力蛋白臂相关的蛋白质,为生成蛋白复合体中的外动力蛋白的重链5结构提供指令,而DNAH5基因的突变可导致无功能的动力蛋白,这种无功能的动力蛋白表现为外动力蛋白臂长度与数量减少,损害运动纤毛功能,进而产生PCD[23-24]。以前的研究[12]报道了儿童PCD相关DNAH5基因的5个热点外显子(34、50、63、76和77)的突变,外显子63是28.0%(37/132)PCD患者中突变最频繁的外显子[25]。本例患儿DNAH5基因突变的外显子为63和61。
PCD的早期诊断对临床治疗及预防长期后遗症非常重要,PCD的诊断依赖于临床表现、鼻呼气NO(nNO)测试、高速视频显微镜分析(HSVA)、透射电子显微镜(TEM)、免疫荧光显微镜分析(IF)和基因检测等[26]。PCD临床表现与其他常见的慢性呼吸道疾病有重叠部分,如囊性纤维化、哮喘等。PCD诊断的4个关键临床特征:①出生6个月内出现全年的持续性湿咳,抗生素治疗未能完全缓解。②出生6个月内出现全年的、每天的、非季节性的鼻窦炎,抗生素治疗未能完全缓解。③足月儿有新生儿呼吸窘迫综合征病史。④内脏转位伴或不伴先天性心脏缺陷[27]。如果其中两个特征存在,则诊断PCD的敏感度和特异度分别为80%和72%,若4项指标都存在,其灵敏度和特异度分别为21%和99%[27]。nNO在成人及超过5岁的儿童中有较好的灵敏度(98.0%)和特异性(99.9%)[28]。PCD患者表现为极低的nNO水平,但囊性纤维化和肺动脉高压疾病也表现为低nNO水平,因此nNO对PCD不是100%特异,但临床上依旧将其作为一种可靠的PCD筛查方法[2]。HSVA具有极好的敏感性(93%)和特异性(93%),但需要区分原发性和继发性异常,对诊断者的经验依赖度较高,因此不能将其作为唯一的检测标准[29]。TEM对PCD的检出率为83%,曾被认为是PCD诊断的“金标准”,但近年来发现部分PCD病例由与超微结构缺陷无关的突变引起而不能被识别,如Nexin-link缺陷,因此TEM不能完全排除PCD[30]。IF能够检测到TEM可识别的所有超微结构异常,以及连接蛋白成分的异常[2]。第二代基因测序技术的使用在PCD等遗传异质性疾病的新基因发现中发挥着重要作用,目前发现有40多个基因可导致这种罕见的疾病,但约30%的PCD潜在的遗传原因尚不清楚[31]。本病例中的患儿未行nNO、HSVA、TEM、IF检测,基因检测结果显示其临床表型符合PCD,突变M1为罕见突变,GnomAD普通人群数据库收录该突变的东亚人群频率为0;突变M2也为罕见突变,GnomAD东亚普通人群数据库未收录,且未见文献报道。患儿妹妹、弟弟基因型相同,临床表型亦符合PCD,依据家长描述患儿妹妹既往检查见内脏转位,可考虑PCD亚型KS。
到目前为止,还没有针对患有PCD患者的具体治疗方案,且临床病例的罕见性和临床表现的复杂性使得制定一个适用于所有患者的标准化管理方案变得困难。PCD治疗的目的是通过早期发现和积极处理并发症来维持或恢复肺功能、预防进行性肺损伤[21],治疗的基石是气道清理,及时治疗呼吸道感染及其并发症。患者应在监测痰培养的指导下,定期进行气道清除技术,以增加黏膜纤毛清除率,并接受防止肺部恶化的抗菌药物治疗[32]。用于促进黏液清除的措施包括正压呼气装置、体位引流、自体引流、手动胸部理疗、“背心疗法”(高频胸壁振荡)等[2]。而肺移植是终末期疾病患者的一种选择,其实行需要制定详细的方案。药物所诱导的提前终止密码子(premature termination codon,PTC)的翻译通读已被证明是减轻遗传疾病症状的有效方法。Bukowy-Bieryllo等[33]测试了不同浓度的氨基糖苷(aminoglycoside,AAG)在与PCD发病机制相关的5个基因中促进PTC突破的潜力,对未来研发治疗PCD的精准药物带来希望。另外一项多国随机对照试验[34]结果表明阿奇霉素维持治疗是减少PCD患者频繁恶化的一种选择,可减少对额外抗生素治疗的需求并预防不可逆的肺损伤。肺康复近年来在临床治疗上被反复提及,可适用于所有的慢性呼吸系统疾病患者,适合儿童的肺康复内容包括呼吸训练、胸科物理治疗和中国传统锻炼等,如缩唇呼吸、主动循环呼吸技术、中医呼吸导引等可在一定程度上帮助肺康复患儿提高运动耐力,降低呼吸困难程度,提高呼吸肌耐受力[35-36]。临床上肺康复应用于PCD患儿的案例较少,随着肺康复研究、实践不断发展、完善,越来越多的PCD患儿可以从中获益。
总之,PCD临床表现多样,与很多呼吸道疾病症状重合,临床诊断困难,但其早期诊断对治疗及预后十分重要。临床上主要以抗感染和对症支持治疗为主,但不能完全根除,因此应重视提高免疫力及进行必要时的手术治疗。近年来遗传学发展迅速,通过基因检测未来可能会确定更多导致PCD的突变基因,随着对纤毛基因及产物的深入了解,基因疗法也给我们带来了纠正纤毛功能障碍的希望。
猜你喜欢
保健医苑(2022年1期)2022-08-30
昆明医科大学学报(2022年2期)2022-03-29
医学研究生学报(2021年4期)2021-12-02
中老年保健(2021年11期)2021-08-22
现代临床医学(2021年4期)2021-07-31
辐射研究与辐射工艺学报(2021年2期)2021-05-06
临床与实验病理学杂志(2021年3期)2021-04-25
中国生殖健康(2020年4期)2021-01-18
实用皮肤病学杂志(2020年2期)2020-05-11
湖北农业科学(2014年11期)2014-09-10
