
一个以高血压、骨折为首发症状的Ⅰ型神经纤维瘤病家系分析
2022-10-13 12:58毛澄源秦小飞单迎光史长河范丽媛董亚丽郑惠敏李心蔚胡正威许予明
郑州大学学报(医学版) 2022年5期
苏 赟,毛澄源,秦小飞,单迎光,史长河,范丽媛,董亚丽,郑惠敏,李心蔚,张 槊,胡正威,杨 靖,许予明
1)郑州大学第一附属医院神经内科 郑州 450052 2)郑州大学第一附属医院心血管内科 郑州 450052 3)河南省肝病药理重点实验室 郑州 450052
Ⅰ型神经纤维瘤病(neurofibromatosis type 1,NF1)为单基因遗传病,是由神经外胚层发育异常而导致渐进性多系统损害的常染色体显性遗传病,发病率为1/3 000~1/2 500[1],临床表型异质性大。美国国立卫生研究院制定了统一的NF1诊断标准[2],至少符合以下两条即可诊断为NF1:①有6个或6个以上牛奶咖啡斑,最大直径≥5 mm(青春期前)/15 mm(青春期后)。②有2个或2个以上任何类型的神经纤维瘤或1个丛状神经纤维瘤。③腋窝或腹股沟区有雀斑。④有视神经胶质瘤或其他脑实质胶质瘤。⑤有2个或2个以上虹膜Lisch结节。⑥骨发育不良或有明确的骨性病变。⑦一级亲属患NF1。
Neurofibromin 1(NF1)基因突变是该病的遗传学病因。NF1基因编码的神经纤维蛋白是原癌基因Ras的负性调控因子,NF1基因突变或异常表达导致神经纤维蛋白功能降低或丧失,从而引起Ras信号通路过度激活,细胞大量增殖且难以凋亡,进而增加各种良性及恶性肿瘤的发病风险[3]。
现对一个以高血压、骨折等不典型表现为首发症状的NF1家系进行报道,并利用目标捕获高通量测序技术[4]对该家系成员进行NF1基因突变检测。本研究经郑州大学第一附属医院伦理委员会批准。
1 临床资料
1.1 先证者资料男性,12岁,汉族,因“血压升高1年”收治于郑州大学第一附属医院。患者1 a前因右侧股骨转子间骨折于外院行骨折固定术后发现血压升高,最高达180/120 mmHg(1 mmHg=0.133 kPa),无头晕头痛、恶心呕吐、视物模糊等;肾上腺增强CT示左侧肾上腺瘤,于外院行手术治疗,术后口服苯磺酸氨氯地平片降压,血压控制于(140~150)/(90~100) mmHg。
近1 a血压持续偏高,伴体位变动后头晕、休息后好转。自述幼年时即出现躯干、颈部及四肢皮肤多发性深棕色牛奶咖啡斑,逐年增大和增多,未诊治。查体:身高150 cm,BMI 20.89 kg/m2;胸腹部呈弥漫大片状牛奶咖啡斑,与周围组织界限清楚(图1);左上肢血压166/108 mmHg,右上肢158/99 mmHg,左下肢155/93 mmHg,右下肢149/94 mmHg;心脏、肺部、腹部及神经系统检查均未见异常。血尿便常规、肝肾功能、血电解质、血脂、炎症指标基本正常。24 h尿儿茶酚胺、24 h尿游离皮质醇、24 h尿醛固酮及24 h尿香草苦杏仁酸结果均正常;促肾上腺皮质激素及皮质醇节律无异常;立卧位肾素活性、血管紧张素Ⅱ及醛固酮水平均正常。24 h动态血压监测:全天平均血压158/93 mmHg,心率86次/min;日间平均血压164/95 mmHg,心率90次/min;夜间平均血压149/87 mmHg,心率84次/min。眼科裂隙灯检查未见虹膜Lisch结节及其他损害。
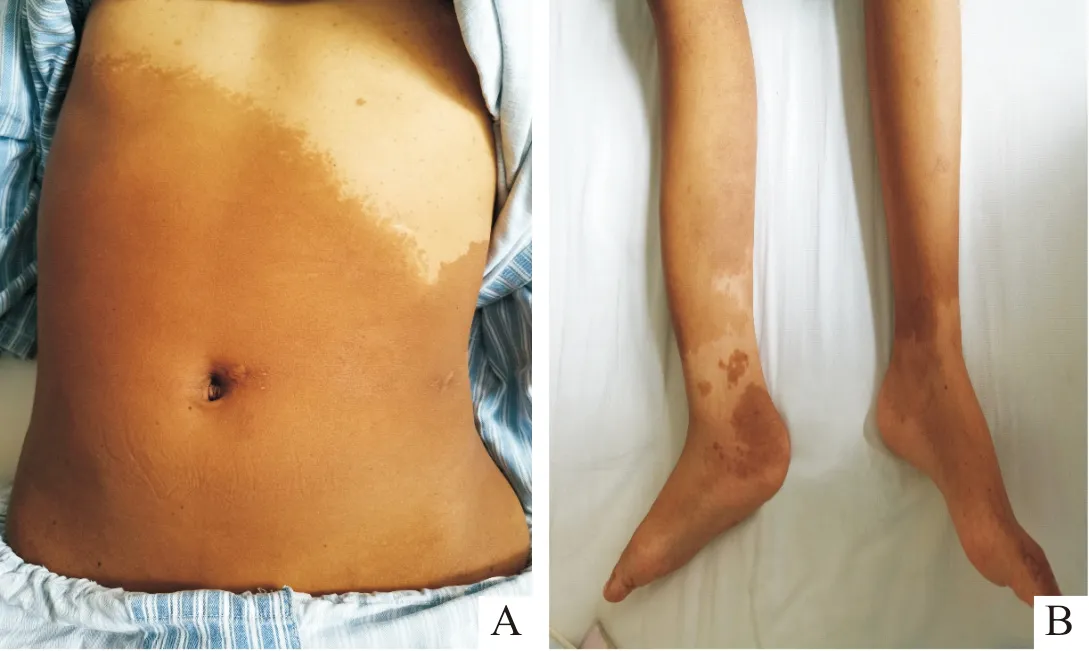
A:胸腹部弥漫大片状牛奶咖啡斑;B:下肢散在牛奶咖啡斑
超声检查示:心内结构及功能未见明显异常,肝胆胰脾未见明显异常,双肾大小形态正常,右肾囊肿,双侧肾动脉走行、内径及血流速度未见异常。CT示:右肾囊肿,右侧肾上腺大小、形态及密度未见异常,且增强后未见异常强化;脊柱旁(约双肾水平)见多发软组织密度影,部分融合成团,右侧为大,边界尚可,密度均匀,增强后未见明显强化,考虑神经来源肿瘤;L3椎体骨质破坏,右侧臀大肌萎缩。腰椎及双髋关节MRI示:双侧骶丛神经增粗,右侧坐骨神经走行区及腰段水平椎间孔神经走行区内多发团片状、串珠状长T2信号(图2),考虑神经纤维瘤;腰椎体形态失常,腰椎后突,腰背部皮下软组织水肿(图2);双侧骶髂关节炎症,右侧股骨近端骨折伴骨髓水肿、周围软组织水肿、滑膜增生。因先证者拒绝未行组织病理学等检查。

A:髋关节T2WI横轴面抑脂图像,见右侧坐骨神经走行区多发串珠状长T2信号;B:腰椎T2WI横轴面图像,见腰段水平双侧椎间孔神经走行区内对称性团块状长T2信号;C:T2WI矢状面图像,见腰椎体形态失常,腰椎后突,L3椎体骨质破坏,腰背部皮下软组织水肿
上述结果不支持继发性高血压的诊断,鉴于既往肾上腺瘤切除术后高血压持续不缓解,予“氨氯地平+非洛地平”口服控制血压,血压控制于140/80 mmHg。请骨科及神经外科会诊,建议定期复查,对症治疗。为明确诊断,进一步行基因检测。
1.2 家系调查该核心家系共2代3人,无近亲结婚,否认高血压家族史。先证者母亲躯干部位密集分布雀斑样咖啡色小斑点,多处散在褐色斑,直径数毫米至数厘米,无高血压及其他伴随症状。先证者父亲无类似表现。
1.3 NF1基因突变检测检测获得受试者知情同意。采用EDTA抗凝管收集先证者及其父母外周静脉血2 mL,采用纯化试剂盒法(北京百泰克,货号DP2201)提取全血基因组DNA,对NF1基因全部编码区外显子进行目标捕获高通量测序,对可疑突变进行Sanger测序验证,将测序结果与UCSC数据库人类基因组信息(GRCh37/hg19)进行比对。测序委托北京金准基因科技有限责任公司完成。高通量测序目标区覆盖度达99.90%,目标区平均测序深度不小于200×,目标区测序深度大于20×的覆盖率达到99.90%。结果显示:先证者NF1基因第38号外显子存在杂合突变c.5719delG。
Sanger测序结果(图4)证实先证者存在NF1基因第5719位碱基G缺失,造成该位置以后出现连续套峰,在蛋白水平将导致第1907位氨基酸由谷氨酸变为赖氨酸,并在之后的第14位形成终止密码,导致蛋白质翻译提前终止,形成仅由1 920个氨基酸组成的截短多肽。其母此位点存在相同的杂合移码突变,而表型正常的父亲不携带此突变,符合家系共分离。

图4 该家系NF1基因突变位点测序图(箭头所示为c.5719delG)
1.4 突变位点致病性分析及蛋白预测检索人类基因突变数据库(The Human Gene Mutation Database,HGMD)专业版2022.1(http://www.hgmd.org/)及外显子组整合数据库(The Exome Aggregation Consortium,ExAC)3.3版(http://exac.broadinstitute.org),参照人类基因突变协会命名规则及美国医学遗传学与基因组学学会遗传变异分类标准与指南[5],参考HGMD数据库报道的NF1转录版本(NM_000267.3),对该突变进行分析。结果显示,HGMD和ExAC中均未检索到该突变位点;中外文数据库中均未检索到该突变致病性的相关文献报道。先证者及其母亲携带可致截短型蛋白的移码突变,为非常强的致病性证据(PVS1:基因存在功能丧失型变异);ExAC数据库中未见该突变在正常对照人群中的频率,为中等致病性证据(PM2:正常人变异数据库未见报道);携带该突变的先证者及其母亲的临床表现符合NF1,为支持致病性证据(PP4:突变携带者的表型或家族史高度符合某种单基因遗传疾病)。因此根据 ACMG指南,该突变被评定为“致病(pathogenic)”。
利用SIFT(http://sift.jcvi.org/)和PolyPhen-2(http://genetics.bwh.harvard.edu/pph2)等生物信息学预测软件对突变蛋白功能进行预测。基于HumanVar数据库,PolyPhen2分值为0.969,表明该突变为“很可能有害(probably damaging)”,导致蛋白结构或功能改变的可能性大。
2 讨论
NF1为单基因遗传病,临床表型异质性大,诊断主要基于临床表现,但典型的特征具有年龄相关性,即牛奶咖啡斑、神经纤维瘤、腋窝或腹股沟雀斑等表现出现时间跨度大,故疾病早期往往由于缺乏特异性而难以明确诊断[6]。本家系中,先证者骨骼病变明显,以右侧股骨近端骨折为首发症状;影像学结果示L3椎体骨质破坏及腰椎后突。神经纤维瘤病导致脊柱畸形的具体机制尚不清楚,推测可能与原发外胚层缺陷及神经纤维瘤组织直接侵入骨骼等有关[7]。1.5%~39.0%的NF1患者伴发椎旁神经纤维瘤[8-10],椎旁神经纤维瘤过度生长可侵犯脊椎,造成椎体骨质破坏及形态失常,进一步导致脊柱结构改变。既往研究[11]发现,约16%的NF1患者合并高血压。国内曾报道过3例NF1患儿合并高血压,均为肾血管性高血压[12-14]。本家系先证者合并高血压病(3级,低危),相关检查均未见明显异常,不支持继发性高血压的诊断,考虑到高血压持续不缓解,故首选药物治疗,稳定控制血压并定期监测;同时加强随访及用药指导,以期预防靶器官损害及延缓心脑血管不良事件的发生,并警惕继发性高血压的发生。此外,先证者的胸腹部呈弥漫大片状、不规则形、光滑的色素异常沉着病损(直径约16 cm),颜色均一,边界清晰。如此巨大的牛奶咖啡斑在神经纤维瘤病患者中鲜有发现,故需收集更多的病例,以探究其是否为一种独特的临床表型。
NF1基因自发突变率高达50%,目前HGMD数据库已收集超过3 700种NF1基因突变,包括单核苷酸替换、插入、缺失及剪切等多种类型的突变[15]。但在已报道的突变中,尚未发现热点区域,也未发现基因型与临床表型之间的相关性[16]。目前研究[17]发现,NF1基因型与表型的相关性仅体现在特定的基因突变类型中,如NF1基因第17号外显子3 bp框内缺失与NF1的典型色素特征有关,但不发生皮肤神经纤维瘤。
目标捕获高通量测序技术相比于全基因组测序更高效快捷[4]。本研究采用目标捕获高通量测序结合Sanger测序,对该家系进行了快速的基因检测,结果为先证者及其母携带相同的NF1基因突变,但两者临床表现差异较大,提示基因型与临床表型关系的不确定性。在同一家系,同样的基因突变也可能有不同的临床表现,说明除基因突变外,NF1表型可能还受到镶嵌或修饰基因的调控[18]。本研究所发现的NF1基因第38号外显子杂合突变c.5719delG为新发突变,在HGMD和ExAC数据库中未有报道。该突变造成开放阅读框发生移码改变,理论上导致蛋白质翻译提前终止,形成仅由1 920个氨基酸组成的截短蛋白(p.Glu1907Lysfs*14),影响蛋白质的功能,最终导致疾病发生。
本研究提示对于仅有高血压等不典型临床症状同时合并皮肤病变的患者,应考虑NF1的可能,系统的临床检查及基因检测有助于明确诊断。本研究发现的NF1突变(c.5719delG)丰富了中国汉族人群NF1基因突变谱,可为该疾病的遗传学诊断提供更多信息。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
临床输血与检验(2022年3期)2022-06-22
实用肝脏病杂志(2022年2期)2022-03-21
天津医科大学学报(2021年1期)2021-01-26
医药前沿(2020年20期)2020-11-10
诊断学(理论与实践)(2020年1期)2020-04-28
三农资讯半月报(2020年2期)2020-03-09
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
医学研究杂志(2015年12期)2015-06-10
