
国产化时间分辨免疫荧光法游离雌三醇检测试剂盒性能验证
2022-10-13 04:17:12宋胜楠王珏
系统医学 2022年15期
宋胜楠,王珏
江苏省人民医院产科,江苏南京 210003
对于出生缺陷疾病,目前尚无有效的治疗手段,通过产前筛查和产前诊断技术在宫内及时发现胎儿染色体数目或结构异常,进行二级预防及干预是出生缺陷防治的重要技术手段。中孕期母血清产前筛查是在孕15~20+6周期间抽取孕妇的肘静脉血(空腹),通过定量测定血清中甲胎蛋白(alphafetoprotain,AFP)、人绒毛膜促性腺激素(human chorionic gonadotrophin,HCG)、游离雌三醇(unconjugated estriol,UE3)浓度值,结合孕妇年龄、孕周、体重、既往妊娠史、有无糖尿病、吸烟等因素,通过风险计算软件评估胎儿罹患唐氏综合征(21三体)、爱德华综合征(18三体)及开放性神经管缺陷(open neural tube defect,ONTD)的风险,根据风险高低决定是否进行下一步产前诊断。根据文献报道胎儿患有21三体时,孕妇血清中游离雌三醇含量平均减少约1/4,胎儿患有18三体时UE3平均水平也会降低[1-2]。本研究按照中华人民共和国卫生行业标准WS/T420-2013《临床实验室对商品定量试剂盒分析性能的验证》要求进行[3],从精密度、正确度、线性范围3个方面,对一款国产化UE3时间分辨免疫荧光定量检测试剂盒进行评估,判断该试剂盒是否能达到实验要求并满足临床需求,现报道如下。
1 材料与方法
1.1 材料
①精密度验证样本采用市售中孕期血清学筛查测定室内质控品(批号:20170401)。正确度验证样本采用国家卫健委临检中心2019年中孕期母血清产前筛查测定室间质量评价冻干品(批号:201911、201921、201931、201941、201951)。以上样本均按照说明书要求复溶后分装保存。线性范围样本来源于2018年10月—2019年4月在江苏省妇幼保健院采集的孕中期血清学筛查临床送检血样,孕周为15~20+6周,血样均在采集后24 h内离心分装血清,并在采样后5个工作日内完成定量检测。检测完成后血清样本置于-70℃医用冰箱保存。在本实验中所有血清样本均提前24 h取出复温至室温并混合均匀[4]。
待性能验证的国产游离雌三醇检测试剂盒(时间分辨免疫荧光法)(批号:8600106450);室内质控品(批号:20170401)。所有试剂均在有效期内使用。
②仪器为AutoDELFIA1235全自动时间分辨仪。
1.2 方法
1.2.1 UE3精密度验证采用本实验室在用的中孕期血清学筛查室内质控品(低、中、高3个浓度)作为精密度测试样本,共行5批实验检测,每批实验对3个浓度样本重复测量3次,每个浓度可获得15个测量值,计算变异系数(coefficient variations,CV),包括批内变异系数(CV批内)、批间变异系数(CV批间)和总变异系数(CV总),与试剂盒厂家声称的CV值进行比较,验证其精密度是否可靠。
1.2.2 UE3正确度验证采用国家卫健委临检中心2019年第一次中孕期母血清学筛查测定室间质量评价冻干品作为正确度测试样本,5个浓度均在医学决定水平范围内。共进行5批实验检测,每批实验对5个浓度样本重复测量3次,每个浓度获得15个测量值。统计分析测量偏移值和参考标准不确定度,按照中华人民共和国卫生行业标准WS/T420-2013[3]的要求判断试剂盒的正确度是否可靠。
1.2.3 UE3线性范围验证在验证范围内,取妊娠36周的孕妇血清作为线性范围验证中的高浓度样本[5],取本实验室临床检测中的极低浓度样本作为低浓度样本。按照表1的比例混合制备不同浓度的6个血清样本。每个样本测量3次,在一批测量中完成。考察测量均值与理论值的线性关系[6]。
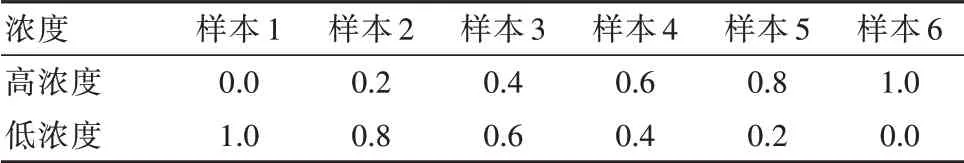
表1 UE3线性范围实验样本制备方法(混合比例)
1.3 观察指标
1.3.1 UE3精密度低、中、高3个浓度样本的CV批内、CV批间、CV总与试剂盒厂家声称的CV值进行比较。
1.3.2 UE3正确度按照中华人民共和国卫生行业标准WS/T420-2013[3]的结果判读规则:当实验室参考物质测量偏移值>参考物质赋值的不确定度,则应将此偏移值的验证区间与参考物质赋值进行比较,如在验证区间内说明差异无统计学意义(P>0.05),应认可本试剂盒正确度验证可靠[7]。
本次验证采用国家卫健委临检中心2019年中孕期母血清学筛查测定室间质量评价冻干品(批号201911、201912、201913、201914、201915),作为参考物质[8]。按照行业标准提供的计算公式,根据国家卫健委临检中心公布的室间质量评价结果,参考物质赋值分别为2.60、4.50、1.80、6.00、6.90,并计算出本参考物质赋值的不确定度。
1.3.3 UE3线性范围 按照比例配置6个理想线性浓度样本后,对其进行检测,以测量均值为横坐标,理论值为纵坐标,拟合曲线方程,得出相关系数,按WS/T2013判读规则,r2>0.995v0即可判断厂家声称的线性范围符合要求[3]。
2 结果
2.1 精密度结果
3个浓度的批内变异系数均小于厂家声称的10.00%;批间变异系数也均0小于厂家声称的15.00%;总变异系数分别为低浓度5.00%、中浓度3.00%、高浓度3.90%,小于厂家声称的总变异系数不超过5.00%。见表2。
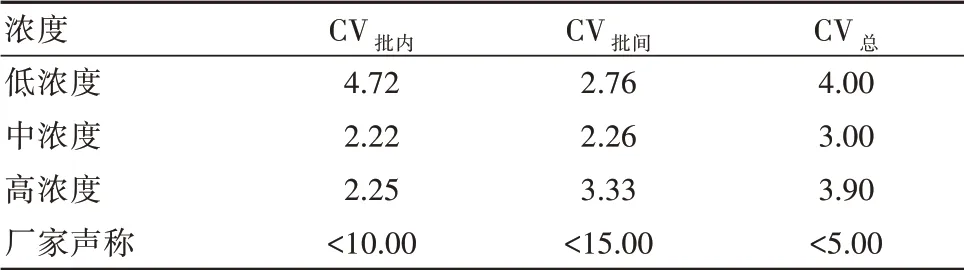
表2 UE3精密度验证批内及批间变异系数结果(%)
2.2 正确度结果
比较后发现,5个参考物质的测量偏移值均大于参考物质赋值的不确定度。因此,继续计算各个测量偏移值的验证区间,结果提示5个参考物质赋值均在测量偏移值的验证区间内。见表3。

表3 正确度验证测量数据(nmol/L)
2.3 UE3线性范围
测量均值和理论值,得出相关系数r2=0.995 4>0.995v0,表明厂家声称的线性范围符合要求。见表4、图1。

表4 UE3线性范围验证血清测量浓度结果
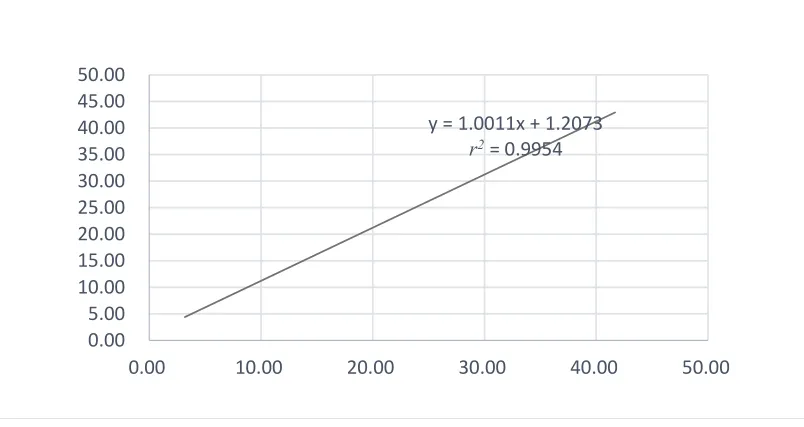
图1 UE3线性范围验证测量值和理论值拟合曲线
3 讨论
本实验室之前一直使用芬兰全进口时间分辨免疫荧光定量UE3检测试剂盒,但由于原材料缺乏,进口试剂停止生产并且完成了国内转产,本院在使用国产化试剂盒之前要按照要求完成国产化试剂盒的性能验证以确保能适用于临床[9-10]。
为对一款国产时间分辨免疫荧光定量UE3检测试剂盒进行性能验证,本实验室从精密度、正确度、线性范围进行评价,了解该试剂盒的分析性能能否达到中孕期母血清产前筛查UE3检测的临床检验要求[11]。采用AutoDELFIA1235全自动时间分辨仪进行检测,验证方法按照中华人民共和国卫生行业标准WS/T420-2013《临床实验室对商品定量试剂盒分析性能的验证》要求进行[13]。游离雌三醇检测试剂盒用于中孕期唐氏综合征筛查,再结合其他指标综合判断胎儿罹患21-三体,18-三体,开放性神经缺陷的风险。临床医生根据不同的的唐筛结果对后续的产前诊断做出不同的判断[12]。
在精密度验证实验中,本实验室选择稳定性良好的市售室内质控品作为测试样本,分别计算低、中、高3个浓度样本的批内变异系数、批间变异系数、总变异系数[12-13]。3个浓度的批内变异系数分别为低浓度4.72%、中浓度2.22%、高浓度2.25%均远远小于说明书声称的10.00%;批间变异系数分别为低浓度2.76%、中浓度2.26%、高浓度3.33%,均小于说明书声称的15.00%;总变异系数分别为低浓度4.00%、中浓度3.00%、高浓度3.90%,小于说明书声称的总变异系数不超过5.00%。按照中华人民共和国卫生行业标准WS/T420-2013判读标准,对比实验室得出的数值和说明书声称的数值得出,验证结果本试剂盒精密度符合要求并能满足实验室临床检验要求。
在正确度验证的实验过程中,关于参考物质的选择,根据WS/T2013建议可首选新鲜冷冻人血清或其他未经加工的人类物质,包括检验医学溯源联合委员会公布的二级参考物质,但是此类参考物质不易获得。故本实验室按照WS/T2013建议,选择大型能力比对或室间质评的样本,即国家卫健委临检中心室间质评样本作为参考物质。参考物质的赋值及不确定度,来源于这次室间质评国家卫健委临检中心公布的数据[14]。
按照行业标准的结果判读规则:当实验室参考物质测量偏移值>参考物质赋值的不确定度,则应将此偏移值的验证区间与参考物质赋值进行比较,根据本次检测结果及统计分析数据,5个参考物质的赋值均在测量偏移值的验证区间之内,说明差异无统计学意义(P>0.05),本试剂盒正确度验证可靠,同时能够满足临床检验需求[15]。
在线性范围验证时,要求所配置的理想线性浓度样本,尽量和临床样本性状一致,因此孕妇样本是线性范围验证的理想样本来源。本院孕妇血清学筛查样本库的采样时间为孕15~20+6周,样本游离雌三醇浓度大多<20 nmol/L,达不到线性范围的高值。故本次性能验证中,选择了妊娠36周的孕妇血样作为接近F点的高浓度样本,在样本库中选择极低浓度的孕妇血样作为低浓度样本,按照比例配置6个理想线性浓度样本,使其均匀分布于试剂盒的标准曲线检测区间。结果显示,本次测量均值与理想值的拟合曲线方程的相关系数符合要求,厂家声称的线性范围符合临床检验要求。
综上所述,本国产游离雌三醇时间分辨免疫荧光定量检测试剂盒,从精密度、正确度和线性范围验证结果显示,其分析性能符合医学实验室临床检验需求。
猜你喜欢
数学物理学报(2022年4期)2022-08-22 04:08:00
中学生数理化·高一版(2021年2期)2021-03-19 08:32:06
World Journal of Clinical Cases(2020年17期)2020-09-18 08:03:24
健康大视野(2020年6期)2020-04-10 06:47:58
检验医学与临床(2020年1期)2020-01-10 04:44:22
医学信息(2019年17期)2019-10-21 06:40:36
中央民族大学学报(自然科学版)(2018年3期)2018-11-09 01:16:34
现代检验医学杂志(2016年5期)2016-08-20 03:17:18
应用海洋学学报(2015年2期)2015-11-22 07:36:40
中国医药指南(2015年10期)2015-10-22 06:22:58
