
17p13.3p13.2微缺失胎儿一例并文献复习
2022-10-10 06:45李燕青傅婉玉吴素霞江矞颖王元白庄建龙
国际生殖健康/计划生育杂志 2022年5期
李燕青,傅婉玉,吴素霞,江矞颖,王元白,庄建龙
17p13.3是一个基因组不稳定区域,与多种罕见神经发育遗传疾病相关,主要包含Miller-Dieker综合征(Miller-Dieker syndrome,MDS)、孤立性无脑回序列(isolated lissencephaly sequence,ILS)、Ⅰ类17p13.3微重复综合征和Ⅱ类17p13.3微重复综合征。分类取决于该区域是否发生了关键基因CRK、PAFAH1B1和YWHAE缺失或重复[1]。研究表明,ILS和MDS患者的基因缺失区域存在相当大的重叠,但MDS患者的缺失通常延伸至17p13.3更末端区域,且具有更典型的临床表型[2]。笔者利用单核苷酸多态性微阵列(single nucleotide polymorphism array,SNParray)检测对1例产前超声及磁共振成像(magnetic resonance imaging,MRI)检查发现颅脑畸形的胎儿进行遗传学诊断及家系分析,明确胎儿为MDS,报告如下。
1 病例报告
患者 女,20岁,因孕25周血清学筛查提示开放性神经管缺陷(open neural tube defect,ONTD)高风险,Ⅲ级彩色超声示胎儿双侧脑室增宽、胎儿脊髓圆锥位置偏低(位于第4腰椎下缘),于2021年6月10日就诊于福建省泉州市妇幼保健院·儿童医院(我院)产前诊断中心。患者孕1产0,妊娠早期因先兆流产口服地屈孕酮片安胎治疗。患者及丈夫平素体健,否认妊娠前及妊娠期有毒有害物质接触史,否认家族遗传疾病史。孕25+5周胎儿颅脑MRI示:胎儿双侧脑室增宽,左侧10.8 mm,右侧12.2 mm。充分告知患者及家属相关风险并签署知情同意书后,于孕26+1周行羊膜腔穿刺术,取羊水细胞进行胎儿染色体核型及SNP-array检测。2021年7月8日(孕29周)羊水染色体核型分析示:胎儿染色体结构及数目均未见明显异常。父母外周血染色体核型分析示:父母双方核型未见异常。SNP-array检测示:胎儿在17号染色体17p13.3p13.2区段存在4.3 Mb片段的缺失[GRCh37]17p13.3p13.2(526~4 388 381)x1(见图1),内含YWHAE(605066)、PAFAH1B1(601545)、VPS53(615850)和BHLHA9(615416)等59个OMIM基因。该片段缺失涵盖MDS疾病区域,临床表型包括无脑回畸形、小头畸形、面部异常、先天性心脏病、发育迟缓及智力障碍等。夫妻双方SNP-array检测结果未见拷贝数异常,提示胎儿17号染色体17p13.3p13.2微缺失为新发突变。本研究已经我院伦理委员会批准(编号:2020No.31)。
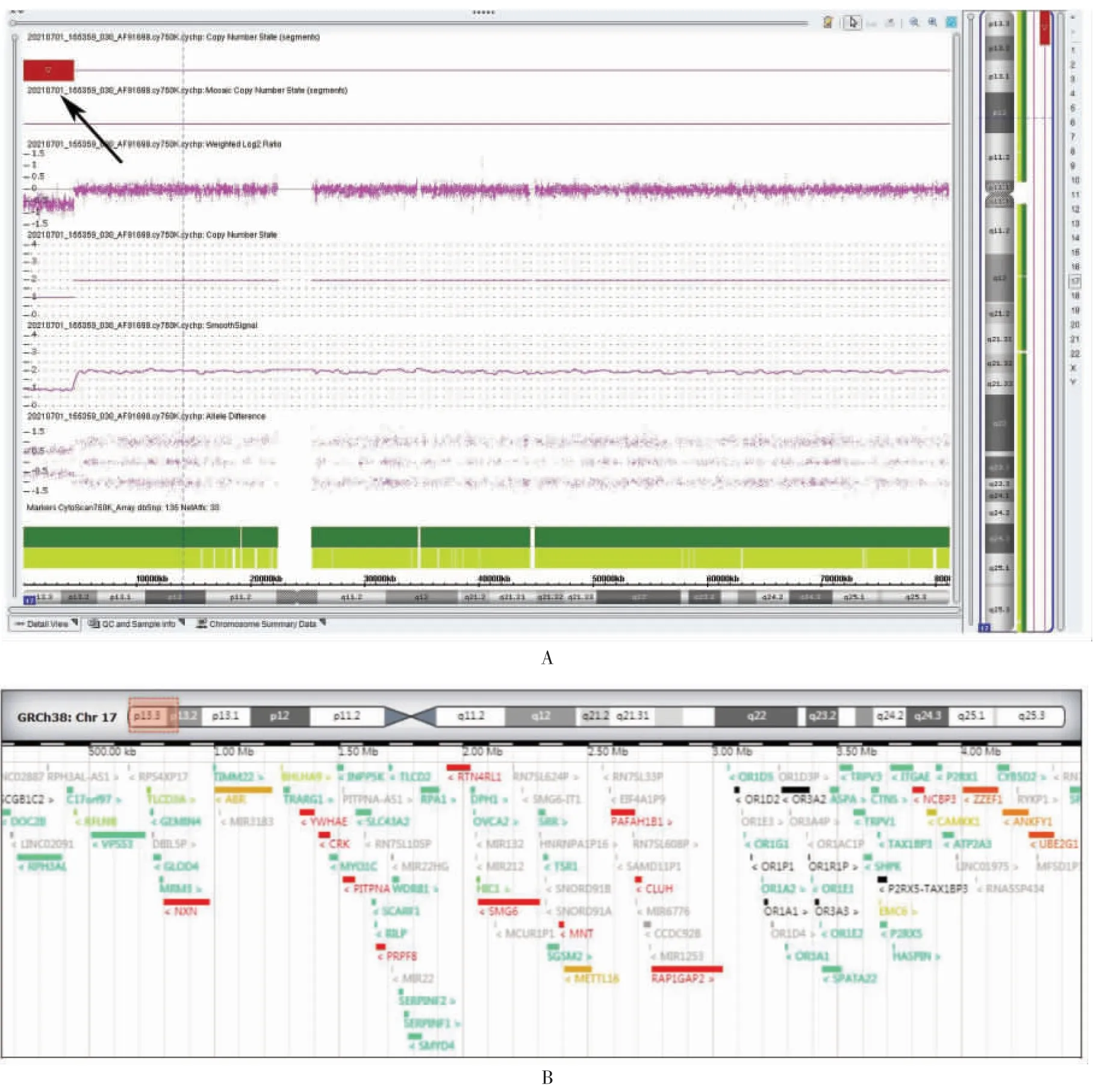
图1 胎儿SNP-array检测结果
孕28+2周彩色超声检查示:双顶径6.74 cm,头围23.72 cm,腹围20.43 cm,股骨径5.03 cm,羊水指数17.57 cm,胎儿双侧侧脑室扩张,胎儿外侧裂池较平直,顶枕沟平直,脑沟脑回显示较少:脑沟脑回发育迟缓,其他疾病待排除,胎儿脊髓圆锥位置偏低。孕28+5周复查胎儿颅脑MRI示:胎儿脑沟、脑裂异常改变,考虑神经元移行障碍——无脑回畸形可能;胎儿双侧脑室增宽(见图2)。于孕29+2周转上级医院复查彩色超声示:胎儿超声孕龄约26+1周,胎儿平滑脑可能,胎儿室间隔膜周部缺损1.8 cm。
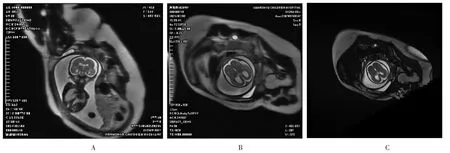
图2 孕28+5周胎儿颅脑MRI图像
经过充分遗传咨询,患者及家属选择终止妊娠,于2021年7月16日孕30+1周给予米非司酮+利凡诺羊膜腔注射引产一男无生机儿,体质量1 060 g,外观未见明显异常,因患者及家属拒绝,未行尸检。
2 讨论
人类染色体17p13.3区段拷贝数变异会导致多种疾病,包括ILS和MDS[3]。MDS的关键基因为PAFAH1B1,有研究认为如果缺失片段同时含有YWHAE基因,往往会导致更严重的无脑回畸形等临床表型[4-5]。与ILS患者相比,MDS患者具有更大的17p13.3片段缺失,其中至少包括PAFAH1B1和YWHAE,通常伴有其他基因[5],其中VPS53基因复合杂合突变与常染色体隐性遗传的脑桥小脑发育不全相关,临床表型包括智力障碍、进行性小头畸形、痉挛和癫痫等[6]。BHLHA9基因纯合突变与常染色体隐性遗传的骨性融合并指联合指骨骨量减少症相关,临床表型包括四肢骨骼发育异常等[7]。因此,MDS患者表现出更严重的无脑回畸形(光滑脑)以及小头畸形和颅面畸形[8]。颅面畸形及胎儿生长受限也是17p13.3微缺失综合征的常见特征,有研究显示这些症状可能与CRK基因单倍剂量不足有关[9]。此外,多项研究发现PAFAH1B1基因以剂量依赖性方式对神经元迁移起至关重要的作用,其单倍剂量不足可导致MDS和ILS[4-5]。而所有MDS的个体都发现了编码14-3-3ε的YWHAE基因缺失,但ILS个体则未受影响[4,10]。Toyo-Oka等[4]通过基因敲除小鼠实验证实14-3-3ε对正常大脑神经元迁移至关重要,14-3-3ε通过与CDK5/p35磷酸化的Ndel1结合并维持Ndel1的磷酸化来调节神经元迁移,这提供了MDS与ILS患者表型严重程度具有明显差异的分子依据。本例患者通过SNP-array检测确诊胎儿在17p13.3p13.2片段存在微缺失,包含MDS疾病区域,表现出典型的无脑回畸形、心脏异常及胎儿生长受限等。
有研究显示,约80%的MDS具有17p13.3区域缺失新发突变,20%是由于父母染色体平衡易位所致[11]。MDS产前常见的影像学表现包括无脑回畸形、胼胝体发育不全、脑室扩大、小头畸形、胎儿生长受限、羊水过多和单脐动脉等[12]。其中,脑室扩大和羊水过多是MDS最常见的超声特征,大多数患者的结构异常超声表现在妊娠晚期被发现[13]。脑皮质发育异常多在孕24周以后才有表现,脑室扩张常是无脑回畸形的线索之一[14]。本例患者于妊娠中期行彩色超声及MRI仅提示胎儿双侧脑室扩张,未见明显结构异常,这可能是由于妊娠中期彩色超声时间较早,未能发现脑结构异常所致。单纯依靠产前超声检查对胎儿神经元移行障碍的检出情况并不理想,MRI拥有多参数、多序列、多平面的成像优势,可弥补产前超声对胎儿神经系统诊断的部分不足[15]。本例患者于妊娠晚期行彩色超声示颅脑异常、室间隔缺损、胎儿生长受限,胎儿颅脑MRI考虑神经元移行障碍(无脑回畸形),基因型与影像学表现相符。MDS的主要临床表型还包括突出的前额、双颞凹陷、短鼻子、鼻孔上翘、上唇加厚、上缘薄朱红色、眼睛间距大、低耳位、小颌畸形等特征性的畸形面部外观[16],但这些特征缺乏特异性,产前难以通过影像学检查发现[17]。除了显著的面部畸形外,MDS有时与其他先天性异常有关,例如肾脏、胃肠道和心脏缺陷[2]。17p13.3缺失的临床表型取决于缺失片段的大小,目前国内报道的相关病例缺失范围从1.4~5.2 Mb不等[11,17-18]。本例胎儿缺失范围较大,累及MDS疾病关键区域,结合超声及MRI结果,胎儿出生后可出现智力低下、难治性癫痫、通常于2岁内夭折[1-3,16],考虑预后不良,经遗传咨询后孕妇及家属选择引产。
综上,由于胚胎发育的特点,胎儿神经系统发育随孕周逐渐完善,孕32~33周胎儿脑沟回的发育基本完成,因此MDS胎儿在不同孕周的影像学表现轻重程度不等。绝大部分确诊的MDS胎儿有2种及2种以上的超声检查异常,少数MDS胎儿仅表现为轻微的超声异常改变[18]。由此可见,临床上利用影像学技术联合SNP-array检测进行胎儿产前遗传学诊断具有重要价值,尤其能够减少致病性染色体微缺失/微重复综合征胎儿的出生,减轻家庭及社会负担。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
锦州医科大学报(2022年3期)2022-06-06
东南大学学报(医学版)(2021年6期)2022-01-27
影像研究与医学应用(2021年15期)2021-09-12
天津医科大学学报(2021年1期)2021-01-26
作文与考试·小学低年级版(2021年2期)2021-01-18
医药前沿(2020年20期)2020-11-10
中国CT和MRI杂志(2020年2期)2020-03-10
三农资讯半月报(2020年2期)2020-03-09
婚姻与家庭·性情读本(2016年5期)2016-05-14
