
基于WGCNA鉴定子宫颈鳞状细胞癌CD8+T细胞浸润相关的生物标志物
2022-09-29 07:33凡志祥李坤张彦彦左世凯黄冬梅
中国现代医药杂志 2022年7期
凡志祥 李坤 张彦彦 左世凯 黄冬梅
子宫颈癌是最常见的妇科恶性肿瘤,其发病率和死亡率在女性恶性肿瘤中位居第四。子宫颈鳞状细胞癌(Cervical squamous cell carcinoma,CSCC)占子宫颈癌的80%~85%,是最常见的子宫颈癌亚型[1]。近年来,肿瘤免疫检查点治疗成为子宫颈癌治疗的研究热点。然而,尚未发现用于子宫颈癌免疫治疗的特异性生物标记物[2]。因此,探索免疫相关生物标记物成为研究子宫颈癌免疫治疗的重要方向。检查点免疫疗法已彻底改变了黑色素瘤的治疗方式,并在肺癌、肾癌和头颈癌中显示出显著的疗效[3~6]。目前,子宫颈癌检查点免疫治疗的试验尚处于早期阶段,而将其作为一种治疗选择主要是因为子宫颈癌是一种病毒驱动的癌症,免疫系统应将其视为异物[7]。现已证实CD8+T 细胞有助于肿瘤适应性免疫,且在大多数实体瘤中,高度浸润的CD8+T 细胞对肿瘤治疗有益[8,9]。且与正常子宫颈组织相比,子宫颈癌中的CD8+T 细胞更为丰富[10,11]。因此,鉴定与CD8+T 细胞浸润相关的新生物标志物将有助于探索免疫浸润机制和丰富免疫治疗。
加权基因共表达网络分析(Weighted gene coexpression network analysis,WGCNA)是一种分析多样本基因表达谱的方法。它可以通过对具有相似表达模式的基因进行聚类,形成模块,并分析模块与特定特征(如患者的临床信息)之间的关系,该算法已广泛用于寻找生物标记物[12]。通过估计转录本的相对子集进行细胞类型识别(CIBERSORT)是另一种用于分析基因表达数据的生物信息学工具,该工具使用反卷积算法量化免疫细胞的组成,已成功用于估计各种癌症中的免疫细胞浸润水平,如乳腺癌和肝癌[13,14]。
为探索肿瘤微环境的特征并确定CSCC 的潜在生物标志物,本研究使用CSCC 的基因表达数据进行WGCNA,并利用CIBERSORT 算法计算样品的T 细胞组成,然后鉴定出与CD8+T 细胞浸润水平相关的关键模块和中枢基因。
1 材料与方法
1.1 材料 通过TCGA 数据库(https://portal.gdc.cancer.gov)获取304 个原发CSCC 样本的基因表达谱,并选择中位绝对偏差前10%的5 605 个基因进行额外分析。从UCSC 数据库(https://xenabrowser.net/)中检索285 例CSCC 患者的临床数据,用于后续分析。
1.2 方法
1.2.1 肿瘤浸润性免疫细胞(Tumor infiltrating immune cells,TIIC)的评估 使用R 包“CIBERSORT”估算TCGA-CSCC 数据集中每个样本的22 种TIIC分数[15]。并选择每个样本中T 细胞的7 个亚型分数作为WGCNA 的性状数据。
1.2.2 加权基因共表达网络的构建 为了解5 605个基因之间的相互关系,并确定基因模块和影响肿瘤免疫细胞浸润的中枢基因,使用R Studio 中的“WGCNA”软件包构建加权基因共表达网络[16]。首先,基于成对基因之间的Pearson 相关系数,将单个基因的表达水平转换为相似矩阵。使用表达式矩阵构造一个分层聚类树来检测异常值。然后根据网络拓扑分析函数计算出无标度拓扑拟合指数作为软阈值功率的函数。接着使用拓扑重叠不相似性度量(TOM)计算具有相似表达谱基因之间的模内连接性。最后采用一种动态混合切割方法将这些基因分成不同的模块,每个模块至少有30 个基因。
1.2.3 构建模块-特征关系 应用Pearson 检验法计算模块特征基因与T 细胞浸润水平之间的相关性,选择P值显著并且相关系数高的T 细胞亚型和模块,定义其为中心模块。为确定中心模块中基因的功能,使用R 包“clusterProfiler”进行富集分析[17]。
1.2.4 hub 基因的识别 根据中心模块中每个基因的模块连通性和临床特征关系选择候选hub 基因。模块连通性定义为基因间Pearson 相关性(Module Membership)的绝对值。临床性状关系定义为每个基因与性状之间Pearson 相关性(Gene Significance)的绝对值。将候选hub 基因的Module Membership设置为>0.8,Gene Significance 设置为>0.4。同时选择中心模块中的所有基因,应用检索交互基因的搜索工具(Search Tool for the Retrieval of Interacting Genes,STRING;https://STRING db.org/)构建蛋白质互作(Protein protein interaction,PPI)网络并寻找中心节点[18]。节点连通性>22 的基因被认为是中心节点。使 用Cytoscape(https:/Cyto-scape.org/)展示网络并使用R 包“VennDiagram”进行Venn 分析以比较PPI 网络中的中心节点和候选hub 基因[19]。
1.2.5 hub 基因的验证 使用TCGA 数据库验证hub基因的免疫相关性。首先,根据CIBERSORT 的计算结果提取出每个样本的CD8+T 细胞比例。使用R 包“ggpubr”计算CD8+T 细胞浸润水平与hub基因表达之间的Spearman 相关性,并使用R 包“ggstatsplot”对结果进行比较。同时检索肿瘤-免疫系统相互作用数据库(Tumor Immune System Interactions Database,TISIDB;http://cis.hku.hk/TISIDB)以确定hub 基因和TIIC 之间的Spearman相关性[20]。最后应用R 包“heatmap”将这些结果以热图形式呈现出来。
1.2.6 免疫因子鉴定 hub 基因和不同免疫因子之间的Spearman 相关性来自TISIDB 数据库,其中包括免疫抑制因子、免疫刺激因子、趋化因子和受体。然后使用R 包“heatmap”构建热图,并使用STRING和Cytoscape 选择与相关免疫因子的平均相关系数大于0.6 的hub 基因构建网络[19]。
1.2.7 hub 基因的预后分析 通过R 包“survminer”找到hub 基因表达量的最优截断值,并将患者分为高表达组和低表达组。然后通过R 包“survival”进行Kaplan-Meier 分析。同时采用log-rank 检验对hub 基因进行单因素生存分析,当P<0.05 时差异具有统计学意义。并将有统计学意义的hub 基因进行多因素Cox 生存分析,鉴别出对CSCC 预后有独立影响因素的基因。
1.2.8 基因集富集分析(GSEA) GSEA 是一种基于基因集的富集分析方法[21]。根据基因表达的中值,将样本分为两组,并进行“c2.cp.kegg.v7.0.symbols”基因集富集分析,以P<0.05 和q<0.05 作为统计显著性指标。使用R 包“ggplot2”和“clusterProfiler”对富集途径进行可视化。在圆图中,本研究只显示富集过程的部分核心基因。
2 结果
2.1 CSCC 的加权基因共表达网络 使用R 包“WGCNA”构建5 605 个基因的共表达网络,并对304 个CSCC 样本进行聚类。为构建无标度网络,选择β=4(无标度R2=0.85)作为软阈值功率(见图1A、1B),并使用动态混合切割技术构建了层次聚类树,其中树上的每片叶子代表一个基因,具有相似表达数据的基因靠一起,形成树的一个分支,代表一个基因模块,共生成10 个模块(见图1C)。

图1 选择合适的β值构造层次聚类树
2.2 识别中心模块并进行富集分析 模块特征基因与T 细胞浸润的相关性见图2A。在这10 个模块中黑色模块与CD8+T 高度相关(r=0.4,P=2.3e-13)。黄绿色模块与未活化记忆性CD4+T 淋巴细胞也有相对较高的相关性(r=0.33,P=4.6e-9)。其他模块与T 细胞之间的相关性小于0.3。将与CD8+T 细胞高度相关的黑色模块作为中心模块,并使用R 包“GOplot”分析该模块中所含基因,以进行通路和过程富集分析。前20 个最高富集项中绝大多数与免疫相关(见图2B),其中前3 个最高富集项为免疫系统的过程(Immune system process)、对刺激的调节反应(Regulation of response to stimulus)和免疫应答(Immune response)。

图2 中心模块和功能
2.3 hub 基因的识别和验证 黑色模块中的高度连接基因被视为与CD8+T 细胞浸润水平相关的潜在因素。基于PPI 网络,应用intera-ction score>0.7和degree>22 的截止值(节点)从黑色模块中确定34 个基因作为中心节点,同时使用Cytoscape将其可视化(见图3A)。根据阈值标准(Module Membership>0.8 和Gene Significance>0.4),从黑色模块中筛选68 个基因作为候选hub 基因(见图3B)。最终选择在以上两种方法中均出现的10 个基因为hub 基因(CCL5、CCR5、CD2、CD3D、CD3E、CD8A、CXCR3、IFNG、PDCD1、PRF1),见图3C。为研究这些hub 基因与CD8+T 细胞之间的关系,使用R 软件分析TCGA-CSCC 数据集中hub 基因的表达数据与CD8+T 细胞的相关性。结果显示,10 个基因的mRNA 表达量与CD8+T 细胞的浸润水平呈显著正相关(所有基因的相关系数最小为0.47),见图4A。作为一个示例,图4B 显示了CD3E 表达量与CD8+T细胞浸润水平的关系。查询TISIDB 数据库,以获得肿瘤浸润淋巴细胞的丰度与hub 基因表达之间的Spearman 相关值。结果显示hub 基因与肿瘤浸润淋巴细胞呈正相关。活化CD8+T 细胞(Act CD8)和效应器记忆CD8+T 细胞(Tem CD8)的相关值最高(见图4C)。此外,以TISIDB 数据库为基础,筛选出33 个免疫因子,这些免疫因子与hub 基因的平均相关性大于0.6。基于hub 基因与33 个免疫因子,又构建了免疫浸润互作网络,以此来探索hub基因的免疫浸润机制(见图4D)。而相关性热图详细展示了hub 基因与不同免疫因子之间的关联性(见图5)。
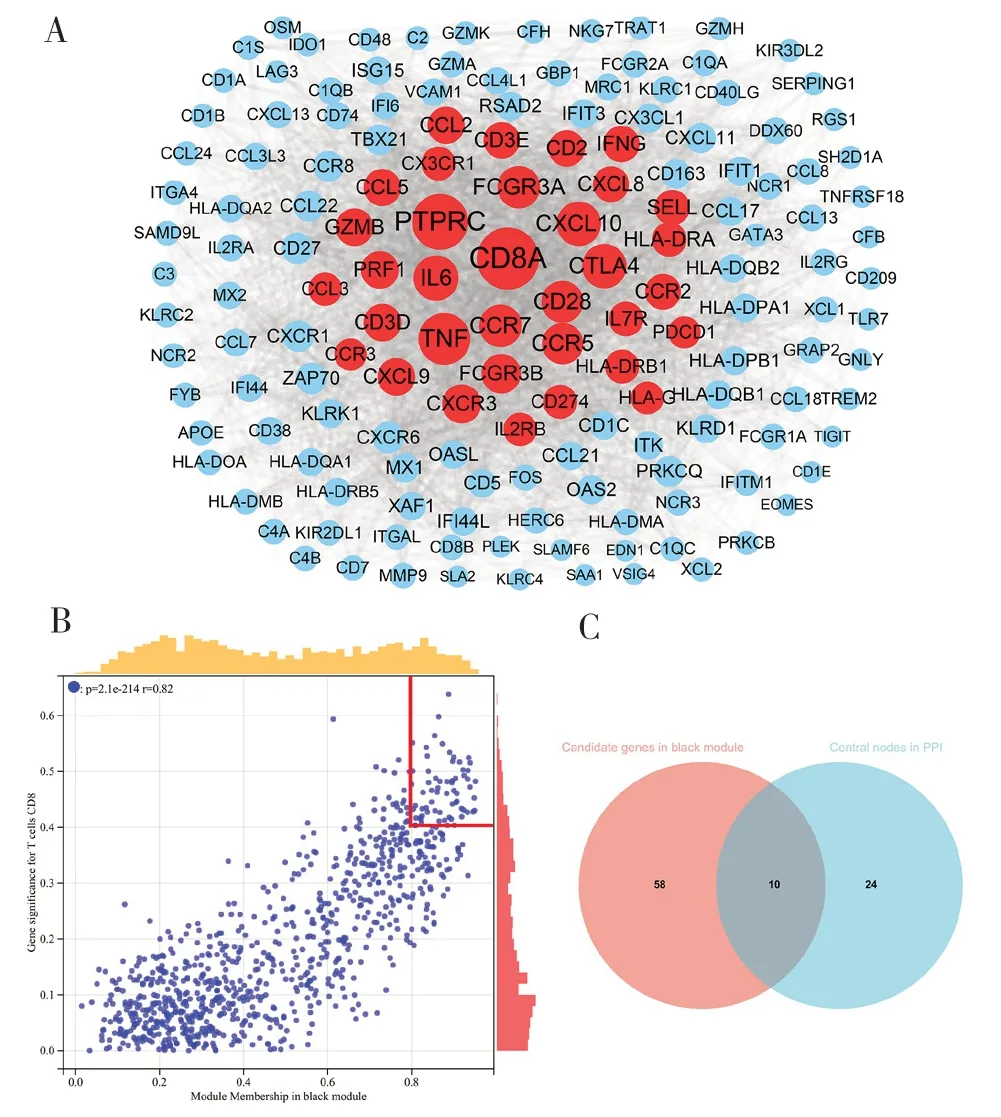
图3 hub 基因的鉴定
2.4 预后生物标志物的鉴定 使用单因素Cox 回归分析探索10 个hub 基因与CESC 患者预后之间的关系,发现除PRF1 与IFNG 外,其余8 个hub 基因均与患者预后有显著的相关性(见图6A)。而多因素Cox 回归分析结果表明,仅CCR5(HR=1.61,P=0.03)与CD3E(HR=0.5,P=0.04)与预后显著相关(见图6B)。Kaplan-Meier 生存分析结果进一步表明,与低表达组相比,10 个hub 基因高表达组患者有更好的总生存期(OS),见图7,这与单因素Cox回归分析结果基本相符。通过观察发现多因素分析结果显示CCR5 为CSCC 的危险因素,这与单因素分析和Kaplan-Meier 生存分析的结果完全相反。因此剔除CCR5 后选择CD3E 作为进一步分析的预后生物标志物。
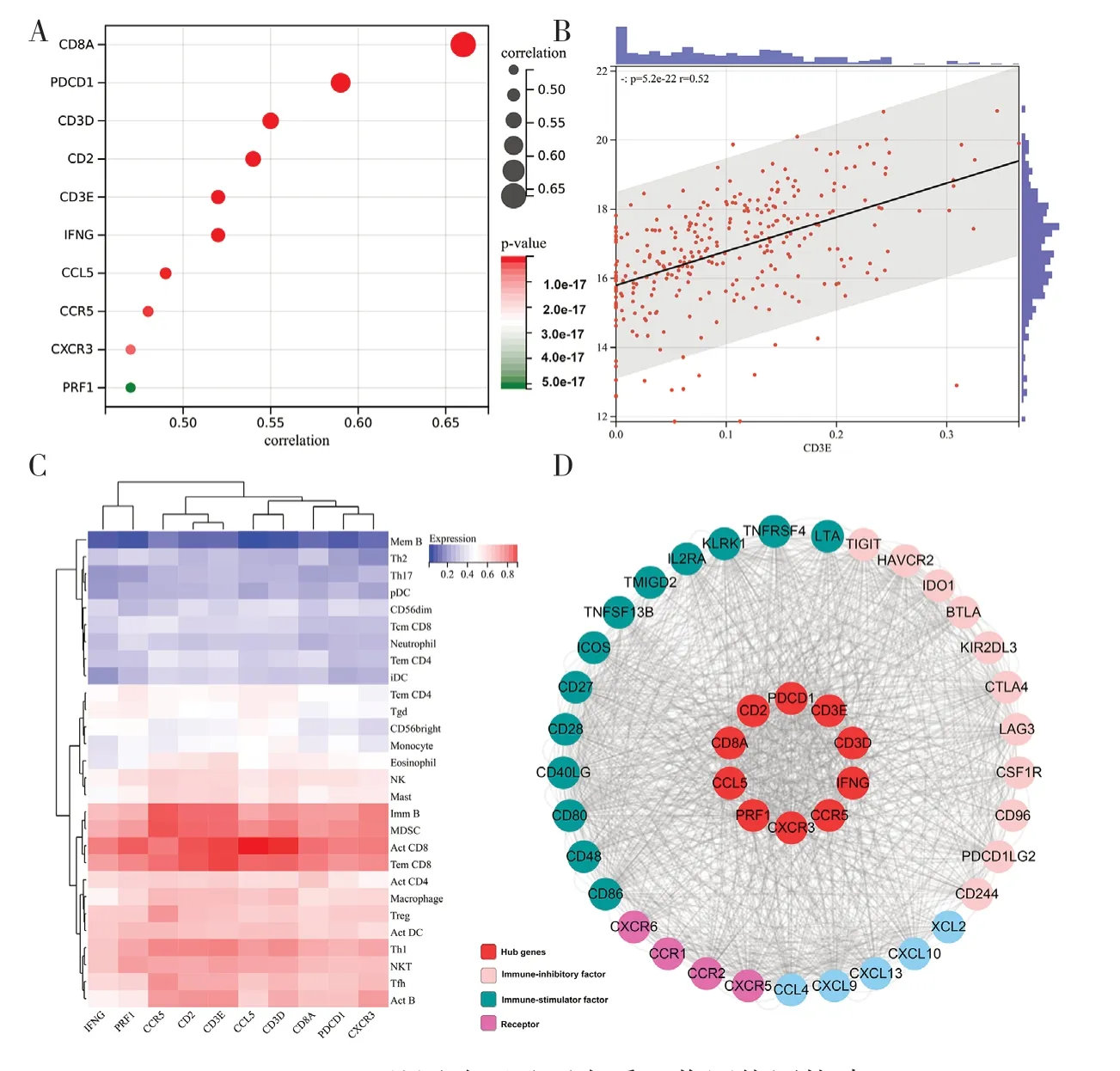
图4 hub 基因验证及蛋白质互作网络图构建

图5 免疫因子和hub 基因之间相关性热图

图6 hub 基因的Cox 回归分析森林图

图7 10 个hub 基的Kaplan-Meier 曲线
2.5 CD3E 的基因集富集分析 根据CD3E 表达中值,将TCGA 中的CSCC 样本分为高表达组和低表达组进行基因集富集分析。结果表明,CD3E高表达组样本富集到的免疫相关通路共21 个(FDR<0.05,q<0.05)。其中4 个富集最显著的途径分别为细胞黏附分子、趋化因子信号通路、细胞因子-细胞因子受体相互作用和T 细胞受体信号通路(见图8A、8B)。而低表达组没有明显的富集途径。
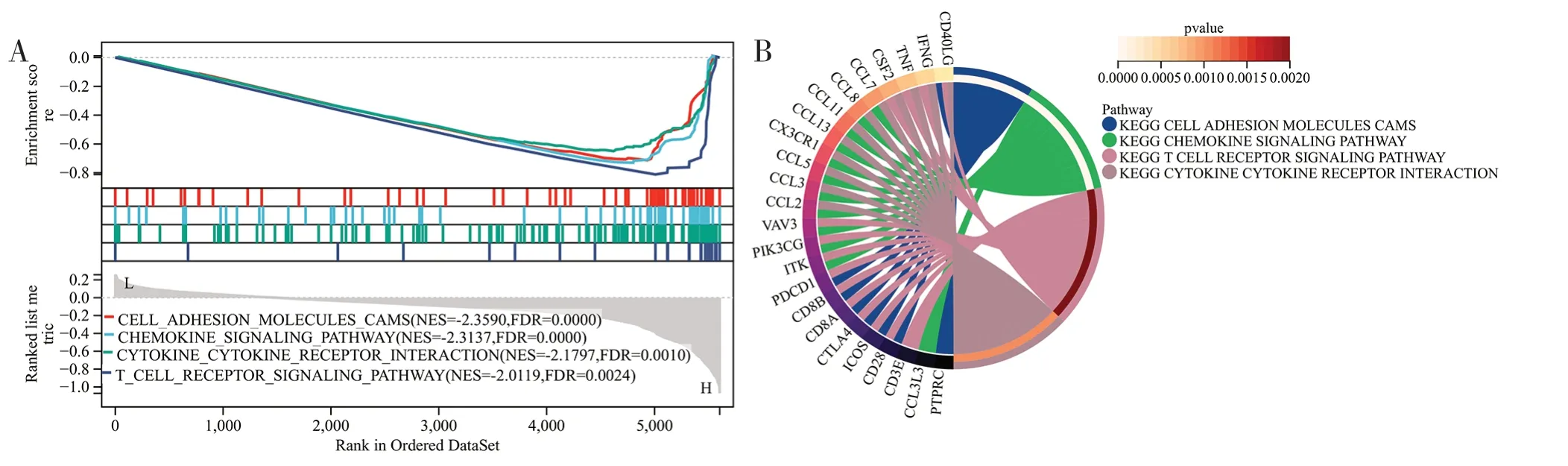
图8 GSEA 富集分析
3 讨论
CSCC 是子宫颈癌的主要组织学亚型,早期CSCC 的预后较好,而对于已发生远处转移的晚期子宫颈癌或复发病例,传统治疗手段的效果并不理想[22]。免疫检查点抑制剂在CSCC 中的成功应用增加了研究人员对探索免疫治疗中潜在特异性免疫相关因子的兴趣[2]。CD8+T 细胞在免疫治疗中起着核心作用。在本研究中,我们鉴定了10 个hub基因,其表达量与CD8+T 细胞浸润水平相关,提示这些基因可能是影响CESC 免疫浸润的潜在因素。在这些hub 基因中,CD3E 被确定为CSCC 潜在的预后生物标志物和治疗靶点。
目前利用动物肿瘤模型、体外细胞系和临床样本技术研究子宫颈癌的分子基础已取得重大进展。然而,CSCC 具有十分复杂的微环境,需要利用更大的数据集来进一步分析。我们使用基因表达矩阵构建共表达网络,计算T 细胞的浸润水平,并确定相关性,以筛选与CD8+T 细胞最相关的基因。对所选模块的基因集富集分析表明,它是一个与免疫高度相关的模块。WGCNA 与PPI 网络之间关联最紧密的基因被认为是hub 基因(CCL5、CCR5、CD2、CD3D、CD3E、CD8A、CXCR3、IFNG、PDCD1、PRF1)。在TISIDB 数据库中查询这10 个基因与免疫细胞之间的关系,发现这些基因的表达与免疫细胞,尤其是CD8+T 细胞显著正相关。以TISIDB 和STRING 数据库为基础,又构建hub 基因和相关免疫因子之间的CD8+T 细胞浸润网络,以探索CSCC的免疫机制。分析结果证实hub 基因与CD8+T 细胞浸润水平密切相关,并在免疫微环境中发挥重要作用。我们还对10 个hub 基因进行了单因素、多因素Cox 回归分析和Kaplan-Meier 分析。其中CCL5、CCR5、CD2、CD3D、CD3E、CD8A、CXCR3、PDCD1 与CSCC 患者预后有显著的相关性,且hub基因高表达时患者的预后较好。而多因素Cox 回归分析仅证实CD3E 低表达是CSCC 不良预后的独立影响因素。结合这三种分析结果,我们选定CD3E为CSCC 患者潜在的预后指标和治疗靶点。
CD3 分子可以与T 细胞受体(TCR)结合,形成CD3/TCR 复合物并介导TCR 信号传导和T 细胞分化[23,24]。由CD3E 基因编码的CD3ε是CD3 的一个亚单位,与严重免疫缺陷有关,经常被用作CD3 抗体的蛋白质靶点[25,26]。研究发现,CD3E mRNA 表达水平较低的头颈部鳞状细胞癌患者复发风险较高[27]。在肝癌、乳腺癌中也观察到类似的结果,即CD3E 的高表达倾向于有更好的预后[28,29]。GSEA 结果还表明,CD3E 的高表达显著富集于基质相关信号通路,如T 细胞受体信号通路和趋化因子信号通路。这些结果表明,CD3E 可能还参与了肿瘤微环境从免疫型向代谢型的转变。因此CD3E 有望成为CSCC 的预后指标和新的免疫治疗靶点。
总之,本研究尝试使用WGCNA 和CIBERSORT算法来识别CSCC 潜在的CD8+T 细胞相关生物标志物,并发现10 个与CSCC 预后相关的hub 基因。通过生物信息学验证,CD3E 被确定为CSCC 免疫治疗的潜在生物标记物和免疫治疗靶点。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
康颐(2020年13期)2020-11-10
家庭医药(2020年10期)2020-10-30
现代养生·上半月(2020年4期)2020-04-20
中国保健营养(2019年4期)2019-09-10
华人时刊(2018年17期)2018-11-19
大众健康(2018年10期)2018-10-22
数学学习与研究(2018年7期)2018-05-16
华东师范大学学报(自然科学版)(2018年3期)2018-05-14
山东青年(2017年11期)2018-03-29
