
HPLC同时测定麦柴方中山麦冬皂苷B和柴胡皂苷A的含量
2022-09-28 07:41蔡碧蓉罗金树
按摩与康复医学 2022年18期
蔡碧蓉,罗金树
(广东省第二中医院(广东省中医药工程技术研究院),广东 广州 510095)
麦柴方经我院消化内科医师临床验方总结、改进而成,通常用于反流性食管炎和慢性胃炎、胃溃疡患者的治疗、辅助症状控制及恢复[1-2]。该方君药为麦冬、柴胡,臣药为法半夏、郁金,其余丹参、厚朴、蒲公英、黄芩等为佐使药。预实验结果表明,该方疗效显著,安全性较好。由于方中药味较多,成分复杂,有必要对其中成分进行筛选和分析,建立多个指标成分含量同时测定的方法,用以评价该方汤剂的质量,更好地服务临床实践和患者需要。
山麦冬皂苷B 是麦冬中的主要指标性成分,具有抑制结肠癌细胞生长,促进其凋亡的功效;同时该成分对于人胃癌细胞也具有较好的抑制作用,可以作为指标性成分对麦柴方进行基于功效主治的质量评价[3-6]。柴胡中有多个皂苷类成分,其中柴胡皂苷A和柴胡皂苷D研究报道较多,且被现行版《中国药典(2020 年版)》列为含量测定指标;文献报道显示,上述成分能够较好地对肝炎和肝癌起到抑制作用,可能是柴胡疏肝解郁、温中行气的物质基础[7-8]。因此,本文选择上述两种成分为指标,建立含量同时测定方法,进而评价麦柴方的质量。
1 仪器和材料
1.1 仪器 Waters2695e 高效液相色谱仪,配备PDA 检测器和自动进样器,购自美国Waters 公司;Mettler Toledo 电子精密分析天平,购自瑞士Mettler Toledo公司。
1.2 材料和试剂 柴胡皂苷A(批号:110777-201912)、柴胡皂苷D(批号:110778-201912)和山麦冬皂苷B(批号:111907-201804)对照品购自中国食品药品检定研究院(国家标准物质网);色谱分析和样品制备用乙醇、乙腈、甲酸购自德国Merck 公司,试验用水为Watson 纯净水。麦柴方所用药材购自广州中芝园药业有限公司。
2 实验方法和结果
2.1 对照品溶液制备方法 分别取柴胡皂苷A、柴胡皂苷D 和山麦冬皂苷B 对照品,精密称取约5mL,置于5mL 量瓶中,加甲醇溶解并定容至刻度,即得上述成分含量均为1mg/mL 的对照品溶液。取上述溶液,精密吸取各1mL,转移至10mL量瓶中,加甲醇稀释并定容至刻度,即得三者浓度均为100μg/mL的混合对照品溶液。
2.2 供试品溶液制备方法 取处方量药材,加水适量煎煮两次,每次60min,即得麦柴方汤剂。上述汤剂纱布滤过后,浓缩至约200mL,转移至250mL 量瓶中,加水稀释并定容至刻度,即得麦柴方标准汤剂。精密吸取标准汤剂1mL,转移至10mL 量瓶中,加乙醇稀释并定容至刻度。所得溶液滤过,取续滤液,即得供试品溶液。
2.3 含量测定方法 吸取混合对照品和供试品溶液,所用色谱柱为Waters Xbridge C18(规格为长25cm,内径0.46cm,填料粒径5μm),流动相为0.2%甲酸(A)-乙腈(B),梯度洗脱程序为:0~5min,5%-5%B;5~10min,5%-25%B;10~20min,25%-75%B;20~25min,75%-5%B;25~30min,5%-5%B,后运行时间5min;流速1.0mL/min,检测波长210nm,柱温30℃,进样量10μL。记录所得对照品和供试品色谱图,见图1。
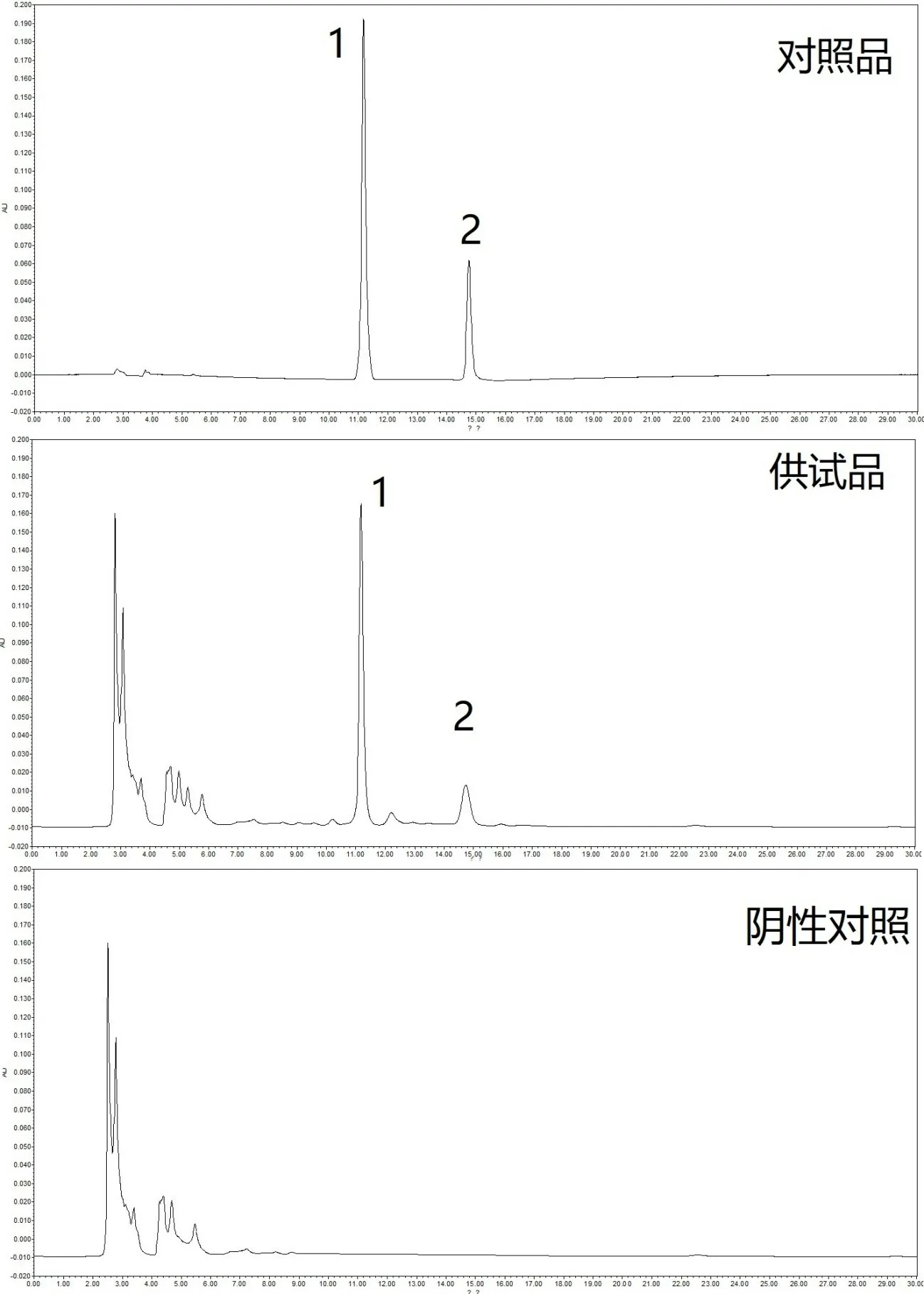
图1 HPLC测定麦柴方对照品和供试品溶液图谱
2.4 方法学考察
2.4.1 标准曲线和线性 取山麦冬皂苷B和柴胡皂苷A 对照品溶液,分别精密吸取适量,转移至多个5mL 量瓶中,加甲醇稀释并定容至刻度,即得上述两种成分含量均分别为5、10、20、50、100、200μg/mL的混合对照品溶液。吸取上述溶液,注入HPLC 中,使用“2.3”项下所述方法分析测定,记录各组样品中山麦冬皂苷B和柴胡皂苷A的峰面积,以峰面积为纵坐标y,对应浓度为横坐标x,使用excel 软件进行线性回归。计算得山麦冬皂苷B的回归方程为y=46355x+44136(r=0.99985),柴胡皂苷A 的归回方程为y=21197x-2979.5(r=0.99975)。上述结果表明,方法线性良好,能够在5~200μg/mL 的浓度范围内对山麦冬皂苷B 和柴胡皂苷A进行准确测定。
2.4.2 精密度考察 吸取供试品溶液,注入HPLC中,使用“2.3”项下所述方法分析测定,重复测定6次,记录各组样品中山麦冬皂苷B 和柴胡皂苷A的峰面积,分别计算二者峰面积的RSD。计算得山麦冬皂苷B 和柴胡皂苷A 峰面积的RSD 分别为1.14%和0.86%,经对比现行版《中国药典》相关规定,上述结果均符合要求,即方法的精密度良好。
2.4.3 重复性考察 按“2.2”项下方法平行制备6份供试品溶液,吸取上述溶液,注入HPLC 中,使用“2.3”项下所述方法分析测定,记录各组样品中山麦冬皂苷B 和柴胡皂苷A 的峰面积,分别计算二者的含量和含量的RSD;计算得山麦冬皂苷B和柴胡皂苷A 含量的RSD 分别为0.65% 和0.29%,经对比现行版《中国药典》相关规定,上述结果均符合要求,即方法的精密度良好。
2.4.4 稳定性考察 取供试品溶液,置于自动进样器中,分别于放置后0、2、4、6、8、12 小时注入HPLC中,使用“2.3”项下所述方法分析测定,记录各组样品中山麦冬皂苷B 和柴胡皂苷A 的峰面积,分别计算二者的含量和含量的RSD;计算得山麦冬皂苷B 和柴胡皂苷A 含量的RSD 分别为1.59%和0.98%,经对比现行版《中国药典》相关规定,上述结果均符合要求,即方法所制备样品在12h内稳定性良好。
2.4.5 加样回收率测定 精密吸取“2.2”项下所制备的麦柴方标准汤剂0.5mL,转移至10mL 量瓶中,按表1、表2 所述最终浓度加入山麦冬皂苷B和柴胡皂苷A 对照品溶液适量,分别配制成低、中、高三个添加浓度的供试品溶液。吸取上述溶液,注入HPLC 中,使用“2.3”项下所述方法分析测定,记录各组样品中山麦冬皂苷B 和柴胡皂苷A的峰面积,分别计算二者的含量、回收率和回收率的RSD。计算得山麦冬皂苷B 和柴胡皂苷A的平均回收率分别为100.31%和99.91%,对应RSD 分别为2.84%和3.32%,经对比现行版《中国药典》相关规定,上述结果均符合要求,即方法准确度良好。
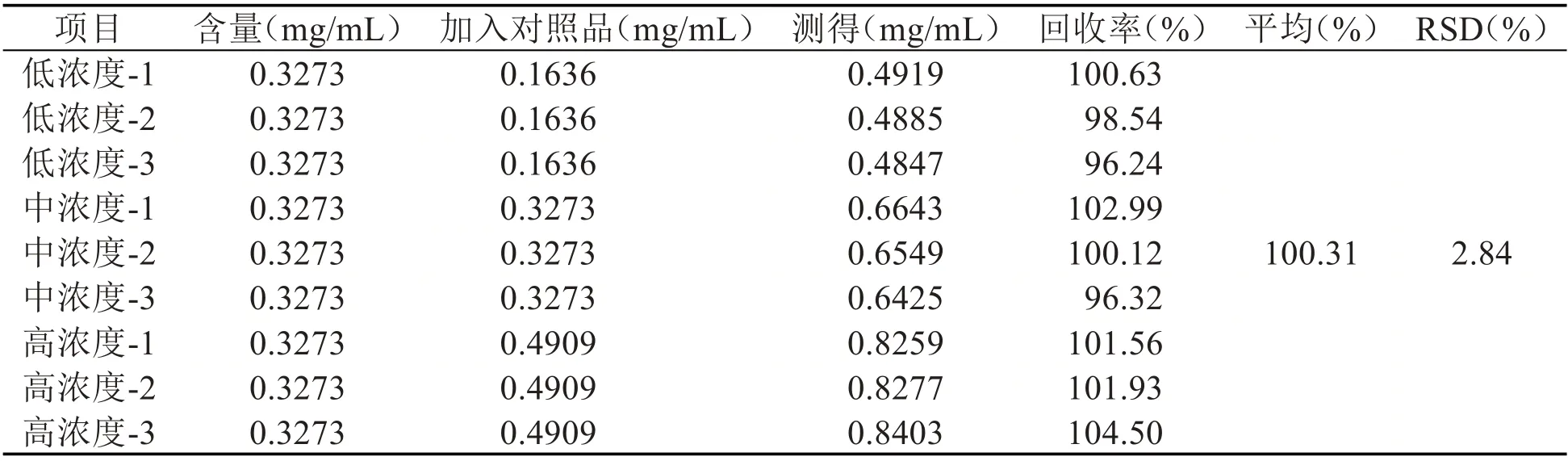
表1 山麦冬皂苷B加样回收率测定结果(n=3)
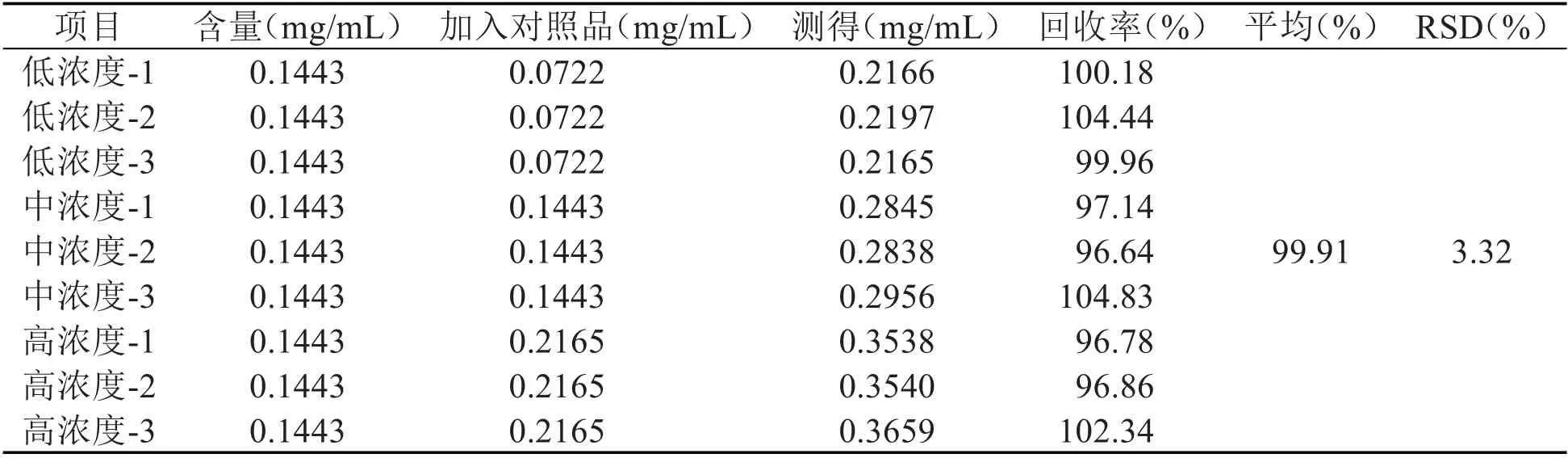
表2 山麦冬皂苷B加样回收率测定结果(n=3)
2.5 样品测定 取处方量药材,按“2.2”项下方法制备供试品溶液,共平行制备10 份,吸取上述溶液,注入HPLC 中,使用“2.3”项下所述方法分析测定,记录各组样品中山麦冬皂苷B 和柴胡皂苷A 的峰面积,分别计算二者的含量。计算得10 批供试品测定结果为,山麦冬皂苷B 含量0.6545~0.7721mg/mL,柴胡皂苷A 含量0.2095~0.3440mg/mL,见表3。
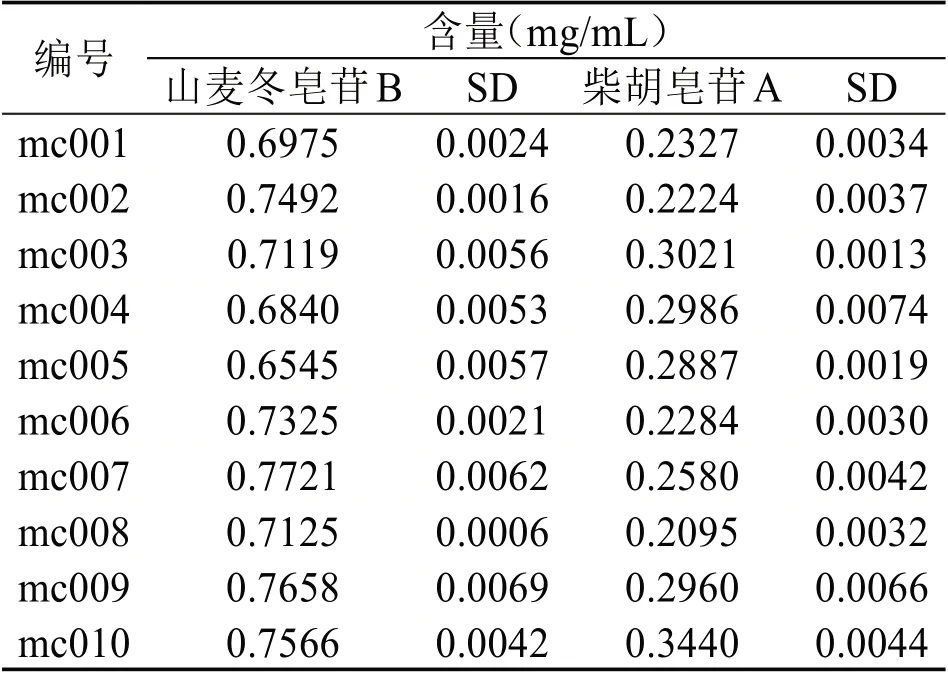
表3 10批样品含量测定结果(n=2)
3 讨论
笔者在预实验中发现,供试品色谱图中并未指认出明显的柴胡皂苷D 色谱峰,但该成分对照品峰较为明显,故基本排除检测方法的问题。经分析和验证,产生上述结果可能是由于使用纯水煎煮药材时,无法有效提取柴胡皂苷D,即麦柴方汤剂中并不含此类成分。综合考虑实验结果和临床使用情况,选择不将柴胡皂苷D 列为含量测定的指标成分。
色谱条件优化和文献调研结果显示,山麦冬皂苷B 和柴胡皂苷A 出峰时间差异较大,两峰附近无干扰峰出现,因此本文“2.3”项下色谱条件能够成功分离二者。影响测定较为明显的因素为供试品制备方法。预实验对提取方式(直接使用乙醇溶解,乙酸乙酯提取后乙醇复溶)、溶剂用量(1:5、1:10、1:20)进行了考察,结果显示乙酸乙酯萃取法提取效率较低,且由于萃取操作较为繁琐,因此重复性和准确度无法保证;溶剂用量考察方面,当溶剂和标准汤剂比例为1:5、1:10 时,前者各指标成分含量均较后者为低(结果略),而当比例变为1:20 时,所得结果较1:10 并无明显变化,表明“2.2”项下所述方法已能够较为充分提取。
猜你喜欢
中国典型病例大全(2022年7期)2022-04-22
中国药学药品知识仓库(2022年5期)2022-04-11
故事作文·高年级(2022年3期)2022-03-17
故事作文·高年级(2022年2期)2022-02-24
农家参谋(2021年7期)2021-10-12
家庭医学(2021年12期)2021-08-23
三农资讯半月报(2020年10期)2020-06-08
小学生导刊(高年级)(2016年10期)2016-10-12
家庭医药(2016年1期)2016-01-20
