
儿童抗MOG抗体合并抗NMDAR抗体双阳性自身免疫性脑炎2例并文献复习
2022-09-27 11:09胡文聪刘秀珍
临床荟萃 2022年8期
胡文聪,刘秀珍
(1.河北北方学院 研究生院,河北 张家口 075000; 2.邯郸市中心医院 儿科,河北 邯郸 056000)
自身免疫性脑炎(autoimmune encephalitis, AE)是由免疫机制介导的中枢神经系统炎性疾病,自2007年抗N-甲基-D-门冬氨酸受体(N-methyl-D-aspartate receptor,NMDAR)抗体脑炎被发现后,随着临床医生对AE的认识和抗神经抗体谱的扩展,确诊病例与日俱增,多种抗神经抗体同时阳性(抗体重叠现象)的AE病例逐渐被报道[1]。其中较为常见的是抗髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein,MOG)抗体合并抗NMDAR抗体双阳性,有学者称之为MOG抗体相关疾病与抗NMDAR脑炎的重叠综合征(the overlapping syndrome of MOG antibody-related disease and NMDAR encephalitis,MNOS)[2]。本研究从临床表现、辅助检查、发病机制、诊疗及预后等方面对邯郸市中心医院收治的2例MNOS患者进行报道并复习相关文献,旨在为该类疾病的诊疗提供更多参考。
1 临床资料
病例1:患儿女,12岁,主因“发热20天”于2020-3-10入院。20天前患儿无明显诱因出现发热,伴有头痛,体温控制后头痛缓解,随后出现性格焦躁,语言偏多,时有说话内容不着边际,说话幼稚,未予诊治。既往体健。入院查体神经系统未见阳性体征,入院后经多次劝说后配合抽血、输液,暂予以抗感染、营养支持等治疗。完善颅脑磁共振成像(magnetic resonance imaging,MRI)示延髓、脑桥、两侧丘脑、中脑导水管周围可见对称性斑片状稍长T1、稍长T2信号影,FLAIR为高信号,磁共振扩散加权成像(diffusion weighted imaging, DWI)未见明显异常高信号,余脑实质未见异常,提示延髓、脑桥、两侧丘脑、中脑导水管周围异常信号(见图1), 考虑①感染性病变?②代谢性疾病韦尼克脑病等不除外。颈椎及胸椎MRI平扫未见异常;腰椎穿刺检查结果(2020-3-12):脑脊液压力正常,蛋白定性阴性,白细胞0.020×109/L,单个核细胞90%,多个核细胞10%;葡萄糖2.33 mmol/L,氯127.6 mmol/L,蛋白0.424 g/L,并将脑脊液及血清外送神经抗体检查。结合脑脊液及颅脑影像学检查结果不能除外病毒性脑炎及中枢神经系统免疫性疾病、遗传代谢性疾病,初步诊断:颅内感染?为进一步明确病因,完善乳酸、血氨、同型半胱氨酸、遗传代谢病氨基酸和酰基肉碱谱分析、尿液有机酸综合分析等检查,均无异常。经抗感染治疗后,患儿体温仍有反复,于2020-3-13予以静点人免疫球蛋白(总量2 g/kg,分5天),患儿体温反复波动于37.2 ℃~37.6 ℃,持续予以抗感染、营养支持治疗,至2020-3-20患儿热峰较前下降,无头痛,同天2020-3-12送检的脑脊液及血清免疫学抗体结果回报:髓鞘碱性蛋白(MBP)0.48 mmol/L,脑脊液抗NMDAR抗体(+)、血抗NMDAR抗体阴性;脑脊液抗MOG抗体(++)1∶3.2,血抗MOG抗体(++)1∶32。结合患儿反复低热,病初伴头痛,疾病进展过程中出现性格、精神、行为改变,语言障碍,遂诊断为MNOS。诊断明确后予以大剂量甲泼尼龙冲击[15 mg/kg,冲击3天停4天,7天为1个疗程,停止冲击期间予以口服泼尼松2 mg/(kg·d),共3个疗程]。自2020-3-31起患儿体温正常,无精神行为异常及语言障碍,完成3个疗程冲击治疗后患儿体温正常,无头痛,精神行为改变恢复正常,出院后3个月随访,患儿无发热、头痛,无精神行为异常,可正常生活及上学。
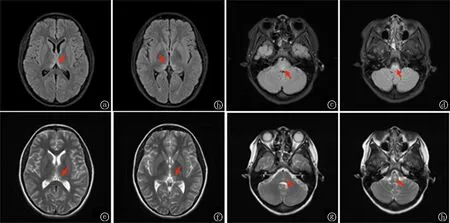
图1 例1颅脑MRI a~d为FLAIR;e~h为T2
病例2:患儿女,10岁,主因“间断抽搐7天”于2022-2-24入院。7天前患儿无明显诱因出现抽搐,意识丧失,面色紫绀,双眼上翻,肢体僵硬,无大小便失禁,持续约10 min;双眼间断向左、向右侧凝视,四肢僵硬,左手抓挠样动作,左足踇趾背伸,无口唇及面色紫绀,可持续3~5 min不等。每次发作均不伴发热。未予特殊诊治。既往无惊厥史,无惊厥家族史。入院时神经系统查体未见阳性体征,暂予以抗感染、降颅压、小剂量甲泼尼龙[2 mg/(kg·d)]抗炎、营养神经等治疗。初步诊断:抽搐原因待查:癫痫?颅内感染?入院后患儿仍有上述表现,伴或不伴有意识丧失,持续时间可长达60 min,遂予以抗感染、降低神经兴奋性、抗癫痫、营养支持治疗,效果欠佳。完善颅脑MRI检查:松果体区见囊状长T2长T1信号,DWI呈高信号,大小约1.4 cm×1.1 cm,边界尚清(见图2),诊断:松果体区良性囊性病变,表皮样囊肿可能。完善性六项、人绒毛膜促性腺激素、盆腔彩色超声均未见异常;腰椎穿刺检查结果(2022-2-26)示脑脊液压力正常,蛋白定性阳性,白细胞0.006×109/L;葡萄糖3.94 mmol/L,氯119.5 mmol/L,蛋白0.262 g/L,免疫球蛋白G(IgG)47.20 mg/L。视频脑电图检查结果提示背景活动慢,弥漫性及广泛性δ波发放,清醒期接近持续发放,后头部及额区著。2022-2-26患儿出现近事记忆障碍(无法回忆上一餐进食的食物)、言语障碍(说话少、语言组织能力差)、意识水平下降(嗜睡),2022-2-28患儿出现行为异常(反复起身、执意下床行走、在床上打转)、不自主运动(张嘴、双手高举)、肢体无力,结合脑脊液结果考虑AE可能性大,遂予以静点人免疫球蛋白(总量2 g/kg,分4天)及大剂量甲泼尼龙[15 mg/kg,冲击3天停4天,7天为1个疗程,停止冲击期间予以口服泼尼松2 mg/(kg·d),共3个疗程]治疗。2022-3-1患儿精神状态较前好转,唤醒后可进行简单词语交流,仍间断有抽搐发作以及行为异常,次数均较前减少。2022-3-3患儿语言功能较前恢复,可成句交流,清醒时间较前延长,出现自主神经功能障碍(尿潴留)、刻板动作(咬唇)、谵妄,仍偶有行为异常(嗓子不自主发声、以头撞击床面)及抽搐发作。同天2022-2-26送检的脑脊液及血清神经抗体结果回报:脑脊液中存在寡克隆抗体;脑脊液免疫球蛋白G 40.30 mg/L阳性(+);抗谷氨酸受体(NMDA型)抗体IgG(血清、脑脊液):阳性(+)1∶30;血清抗MOG抗体: 阳性1∶10。结合患儿临床表现、脑脊液结果,故诊断为MNOS,遂继续按计划完成足量人免疫球蛋白、大剂量甲泼尼龙冲击治疗,加用左乙拉西坦抗癫痫治疗,2022-3-5患儿体温波动在37.5 ℃左右,可自行降至正常,考虑为中枢性发热。经上述治疗,患儿病情好转,清醒时间延长,抽搐发作控制,不自主运动减少。2022-3-14患儿病情反复,意识水平再次下降、行为异常较前增多,遂再次予以人免疫球蛋白(1 g/kg,1天)治疗。自2022-3-15起患儿精神状态逐渐好转,语言组织及表达能力逐渐恢复,抽搐发作消失,不自主运动、精神及行为异常减少。复查视频脑电图:背景活动慢,清醒期可见δ、θ混合慢波发放。出院时近事记忆力仍稍欠佳,语言功能尚可,无癫痫发作及精神行为异常。出院3个月后随访,患儿已能正常上学及生活,无精神行为异常及抽搐等表现。
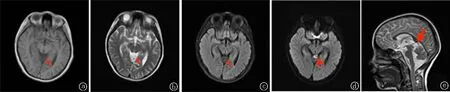
图2 例2颅脑MRI a为T1;b为T2;c为FLAIR;d为DWI;e为矢状位
2 讨 论
MOG抗原仅表达于中枢神经系统少突胶质细胞质膜上,参与髓鞘的黏附,维持髓鞘的完整性[3]。儿童MOG-IgG相关疾病在4~8岁患儿中主要表现为脑病,出现意识水平下降、认知障碍或精神行为异常等症状,在13~18岁患儿中以视神经炎更为多见,可有部分患儿表现为癫痫发作[4-5]。NMDAR通道参与许多复杂的生理和病理机制,可以贮存大量信息,被认为是学习和记忆的基础[6]。在儿童中,抗NMDAR脑炎是最容易识别的AE[7],行为和言语问题、癫痫发作、异常运动是较为常见的早期症状[8],还可伴有近事记忆力下降、自主神经功能障碍等[9],偶尔可出现脱髓鞘病变[2]。下面将从临床表现、辅助检查、发病机制、诊疗与预后等方面对MNOS进行讨论。
2.1临床表现 AE是MNOS最主要的临床表型,抗MOG抗体和抗NMDAR抗体均可引起AE和脱髓鞘病变[10],两者引起的病理变化既可以同时也可以先后出现。本研究报道的2例,年龄分别为10岁和12岁,符合既往研究“该病多见于18岁以下的青少年”的结论[2,11-14]。本研究2例均临床表现既有抗MOG抗体相关疾病的脑病表现,也有抗NMDAR脑炎的表现,且以抗NMDAR脑炎的表现更易被识别。与Hou等[12]提出的MNOS的临床表现与抗NMDAR脑炎重叠更多的观点相符。李清晨等[13]提出,MNOS的临床表现与单纯的抗MOG抗体相关疾病相比,除头痛、发热等脑病症状外,与脑炎相关的症状,如精神行为异常、视力下降、意识水平下降、癫痫发作、肢体运动/感觉障碍、眼肌麻痹、中枢性低通气等,也较为常见,而这些症状通常比典型的抗NMDAR脑炎要轻。同时Zhang等[15]提出,与单纯抗NMDAR脑炎相比,MNOS患者抗NMDAR脑炎症状更少而脱髓鞘症状更多,前驱感染现象、复发病程更为多见。本研究仅对2例MNOS患者进行报道,并未将其与单纯抗NMDAR脑炎或MOG抗体所介导的中枢神经系统脱髓鞘患者进行比较,未能对上述规律进行辅证。但当患者同时或先后出现抗MOG抗体和抗NMDAR抗体所介导的临床表现时,应积极完善全面的神经抗体检查。
2.2辅助检查
2.2.1脑脊液、血清 本研究2例患儿脑脊液均有白细胞轻度升高,蛋白质、糖、氯化物无规律性改变,与既往研究报道的病例检查结果相似,即此类患者脑脊液检查白细胞呈不同程度升高,蛋白质定性可随病程进展而改变,但脑脊液葡萄糖及氯化物的含量变化无明确规律[12-14]。遂当临床所遇病例考虑中枢神经系统感染,且病因不明确时,万不可因脑脊液白细胞数目的轻度增高而误诊为病毒性脑炎进行治疗。本研究2例患儿均在脑脊液或血清标本中检测出致病性神经抗体,为确诊疾病不可或缺的条件。有学者提出,较高的持续性抗MOG抗体滴度的患者复发可能性更高;较高的持续性抗NMDAR抗体滴度更可能导致神经系统后遗症[12]。本研究2例患儿虽然2种抗体滴度不同,但预后均良好,无反复。与Hou等[12]观点不同,却与曹丽平等[14]观点相符,即抗体滴度是否影响预后尚未明确。
2.2.2神经影像学 本研究病例1同时存在幕上及幕下病变,且颅脑MRI提示延髓受累;病例2仅有幕上病变,与既往研究报道一致[2,12-13,16-17],即MNOS患者颅内病变大多位于幕上,部分合并幕下病变。这对临床诊断有辅助意义,因单纯抗NMDAR脑炎患者颅脑MRI可无明显异常,或者仅有散在皮质、皮质下点片状FLAIR和T2高信号,少数病例兼有中枢神经系统炎性脱髓鞘病的影像学特点,大脑白质或者脑干受累[9]。而抗MOG抗体相关疾病患者颅脑MRI病变以两侧脑室旁白质区病灶多见, 皮层、丘脑、海马病灶具有相对特异性, 亦可见于胼胝体、内囊、脑干、小脑[4,16]。
2.2.3神经电生理 目前关于MNOS患者脑电图特点的研究甚少。本文所报道的病例2脑电图示背景活动慢,弥漫性及广泛性δ波发放,清醒期接近持续发放,后头部及额区尤其明显,其结果与抗NMDAR脑炎典型脑电图重合较多[9]。研究报道, MNOS患者脑电图大多数为非特异性慢波,少数为癫痫样放电[12-13]。考虑到抗NMDAR脑炎脑电图偶可见癫痫样放电,故认为抗NMDAR脑炎可能对MNOS患者的脑电图影响更显著。
2.3发病机制 MNOS的发病机制尚不明确,目前讨论最多的是免疫机制,肿瘤(常见的是卵巢畸胎瘤)是已知的抗NMDAR脑炎最常见的免疫触发因素之一。本研究报道的2例患儿发病前无前驱感染史,入院后完善检查均未发现肿瘤,既往病例报道只有极少数患者合并有卵巢畸胎瘤[2, 12-13, 16-17]。故考虑肿瘤与MNOS的发生无直接关系。有学者猜测MOG、NMDAR这两种自身抗原表面可能同时存在于少突胶质细胞,在病理过程中自身系统可能错误地攻击自身抗原,从而产生抗体[18]。MNOS的具体发病机制有待进一步探究。
2.4诊疗及预后 一线治疗为激素、免疫球蛋白、血浆置换;二线治疗为免疫抑制剂。其中激素与免疫球蛋白是主要的一线治疗,二线治疗多用于复发或者对一线治疗效果不佳的患者。本研究2例患儿对大剂量甲泼尼龙冲击及人免疫球蛋白治疗反应良好,无论是脑病症状还是抗NMDAR脑炎的症状均得到很好的控制,且3个月后随访均未复发或遗留后遗症。关于MNOS的病例报道发现,多数患者对大剂量甲泼尼龙联合免疫球蛋白,小剂量激素维持并逐渐减量的治疗方法反应良好,少部分病例出现复发,且复发后再次予以大剂量甲泼尼龙冲击、人免疫球蛋白,亦或是免疫抑制剂治疗均可得到很好的控制,多数预后较好,不遗留后遗症[2,12,16-17]。而杨丹等[16]报道了1例因出现中枢性低通气、呼吸衰竭, 最终临床死亡的病例。提示,大多数MNOS患者对免疫治疗反应良好。在免疫治疗疗程方面,大多数学者认为MNOS患者应采取更长疗程的免疫治疗[1,13],考虑与MOG抗体相关脱髓鞘疾病有较高的复发倾向且可能遗留后遗症有关[19-20]。
综上,MNOS多见于18岁以下青少年,当患者同时或先后出现抗NMDAR脑炎症状、脱髓鞘事件时,则需警惕MNOS的可能性。辅助检查除脑脊液、血清神经抗体外,脑脊液中白细胞数目、颅脑影像学检查、视频脑电图均对该类疾病有辅助诊断意义。多数病例对甲泼尼龙、人免疫球蛋白治疗反应良好,预后尚可,可适当延长免疫治疗疗程,以避免复发或遗留后遗症,但关于其发病机制仍需进一步探讨。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
健康博览(2022年5期)2022-05-24
现代临床医学(2022年1期)2022-02-12
祝您健康(2022年2期)2022-01-14
科教新报(2019年26期)2019-09-10
家庭科学·新健康(2019年1期)2019-03-06
家庭医学(2017年12期)2018-01-15
家庭医学(2014年6期)2014-09-11
为了孩子(孕0~3岁)(2001年3期)2001-06-13
祝您健康(1998年9期)1998-12-25
