
复合物TEMPO…XY中自由基卤键的性质及协同效应
2022-09-20 02:05郭玉琳单爱婷张利利张雪英
河北师范大学学报(自然科学版) 2022年5期
郭玉琳,单爱婷,张利利,张雪英
(河北师范大学 化学与材料科学学院,河北省无机纳米材料重点实验室,河北 石家庄 050024 )
近年来,分子间或分子内非共价相互作用的研究取得了快速发展,非共价相互作用在超分子化学、晶体工程、材料科学及生命科学等领域发挥着非常重要的作用[1-2].卤键是实验和理论研究最深入的非共价相互作用之一,是指卤族元素充当路易斯酸,与含孤对电子或π电子的路易斯碱之间发生的相互作用[3].Clark等[4]指出卤键的形成与卤原子周围电子密度的各向异性分布有关,可以用“σ-hole”概念来解释,σ-hole是指一个空的σ*反键轨道上正的静电势区域,通常位于一个共价键的矢量方向上.卤键具有明显的方向性,静电、极化、色散及电荷转移等起着一定的作用,在晶体工程、分子识别和催化反应中有着广泛的应用[3,5-6].
氮氧自由基是一类含碳、氮、氧等元素以及自旋单电子的有机化合物,性质较稳定且具有特殊的生物活性和磁学性质,可用来作为信号传递的官能团,用于研究药物和其他生物大分子的相互作用[7-8].氮氧自由基还可以作为电子给体与氢键、卤键供体等发生相互作用,Morishima等[9]首次报道了溶液中CH3X与自由基(Me)2NO之间存在卤键作用.Mugnaini等[10]用电子顺磁共振(ESR)能谱法测定了溶液中脂肪族和芳香族碘氟烃与氮氧自由基之间的卤键复合物.Alkorta等[11]定性和定量地分析了在剑桥结构数据库中有关氮氧自由基参与形成的晶体结构中的氢键.一些氮氧自由基化合物还可作为自旋探针用于探测溶液中的氢键和卤键[12].Boubekeur等[13]首次报道了应用卤键控制由4-氨基-TEMPO和1,4-二碘四氟苯组装的一维有机自旋系统.研究发现,卤键可以实现单分子阵列的构建,从而控制有机化合物中特定的相对自旋方向来构建新的分子磁性材料,在晶体工程中是一种可预测的超分子工具.为了进一步阐明卤键在调节磁性能中的作用,Pang等[14]由BTEMPO和DITFB构建了新的磁性共晶,结果表明,卤键不仅可以影响磁性偶联,而且可以利用不同的卤键供体获得不同的磁性.科研人员[15-16]还采用理论计算方法研究了氮氧自由基参与形成的氢键及卤键的性质,但有关氮氧自由基参与形成的非共价相互作用的研究还比较少.本工作中,笔者选取典型的氮氧自由基、TEMPO作为路易斯碱,一系列卤化物XY(X=Cl,Br,I;Y=CH3,CF3,CN)作为卤键给体,对复合物进行量子化学计算.从稳定构型、结合能、电子密度拓扑性质等方面,探究自由基卤键作用的特征;分析双分子复合物TEMPO…XY中取代效应对卤键强度和性质的影响;构建2类三分子复合物NCX…TEMPO…XCN和TEMPO…XCN…XCN,研究卤键间的协同效应.
1 计算方法
密度泛函M06-2X方法[17]能较好地描述非共价相互作用,常用于研究分子间和分子内的相互作用体系[18-19].因此,笔者在M06-2X/Def2-TZVP理论水平上,使用Gaussian 09程序[20]对单体、双分子及三分子复合物进行构型优化和频率计算,确认优化后的几何构型与势能面上的最小值相对应,没有虚频.分子间的结合能是复合物能量与单体能量之和的差值,其中单体构型为单独优化的稳定构型,并且对结合能进行基组重叠误差[21]和零点能(ZPE)矫正.在优化构型的基础上,使用 Multiwfn软件[22]和VMD程序[23]对单体和双分子复合物进行静电势(MEP)和分子生成密度差(MFDD)分析.根据分子中原子的量子理论(QTAIM)[24],使用AIMAll程序[25]计算分子间相互作用的电子密度拓扑性质.
2 结果与讨论
2.1 单体的静电势分析
分子静电势(MEP)是预测许多非共价相互作用的方向和强度的有用工具.图1给出了在电子密度为0.001 a.u.的等值面上TEMPO及卤化物CH3I,CF3I,ICN的MEP图,其他卤化物的静电势分布情况相似.图中TEMPO分子在氧原子周围呈现蓝色区域,其静电势为负值,蓝绿色圆点表示静电势极小值(Vs,min)的位置,数值为-155.9 kJ/mol.对于CH3I,CF3I,ICN分子,在C—I键的反向延长线碘原子的外部存在红色的“σ-hole”区域,橙色圆点表示静电势的极大值(Vs,max)位置.卤化物表面σ-hole处Vs,max列于表1中最后一列,其数值随着X=Cl,Br,I的电负性减小的顺序逐渐增大.当卤原子相同时,随着取代基CH3,CF3和CN的吸电子能力逐渐增强,卤原子处的电子密度逐渐减小,静电势的数值越大.根据单体的MEP图可以预测,TEMPO分子中的氧可以与卤化物的σ-hole区域发生相互作用,形成自由基卤键复合物.
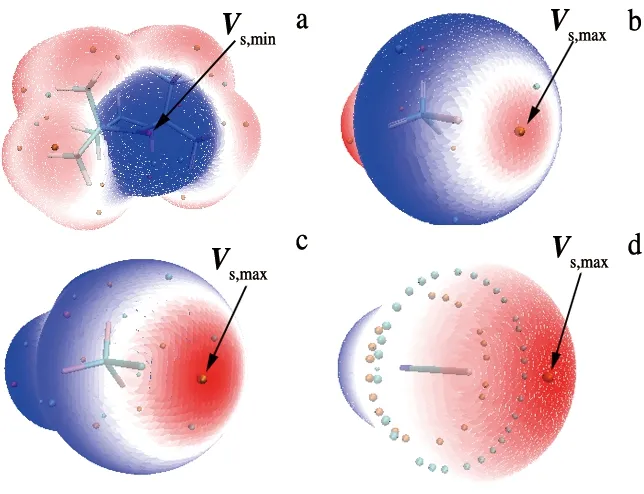
a.TEMPO; b.CH3I; c.CF3I; d.ICN.图1 分子在0.001 a.u.等值面上的静电势图Fig.1 MEPs Maps on the 0.001 a.u. Contour of the Molecular Electron Density
2.2 双分子复合物的构型和能量分析
表1给出了双分子复合物TEMPO…XY(X=Cl,Br,I;Y=CH3,CF3,CN)中的主要构型参数、分子间结合能及卤化物分子σ-hole处的静电势极大值.表中Δd(O…X)表示TEMPO分子中氧原子与XY中卤原子之间的距离与相应原子的范德华半径之和的差值,均为负值,表明分子间发生了相互作用.当取代基相同时,Δd(O…X)的绝对值随着X=Cl,Br,I电负性的减小而逐渐变大;当卤原子相同时,随着取代基CH3,CF3,CN吸电子能力增强而逐渐变大.Δd(C—X)表示复合物中碳卤键的键长相对于单体卤化物的键长变化,可以看出,除TEMPO…BrCN和TEMPO…ICN外,其他复合物中C—X键长变化很小.Δd(N—O)表示复合物中TEMPO中N—O键长相对于单体的键长变化,可知卤键作用对氮氧键键长无影响.
从分子间结合能(ΔE)数据可以看出,当取代基相同时,ΔE的绝对值按照X=Cl,Br,I的顺序逐渐增大;当卤原子相同时,ΔE的绝对值随着CH3,CF3,CN吸电子能力增强而增大.卤键作用强度的变化顺序与Δd(O…X)变化规律一致,卤键越强,分子间相互作用距离越短,Δd(O…X)变化越大.例如,复合物TEMPO…ClCH3中卤键作用最弱,结合能为-2.6 kJ/mol,Δd(O…X)绝对值最小,为0.023 9 nm;复合物TEMPO…ICN中卤键作用最强,结合能为-36.6 kJ/mol,Δd(O…X)绝对值最大,为0.073 0 nm.另外,比较分子间结合能与卤化物σ-hole处静电势极值可以看出,Vs,max数值越大,ΔE绝对值越大,卤键作用越强.卤键结合能ΔE和静电势极大值Vs,max之间具有线性关系,表明静电力在自由基卤键中占有重要地位.
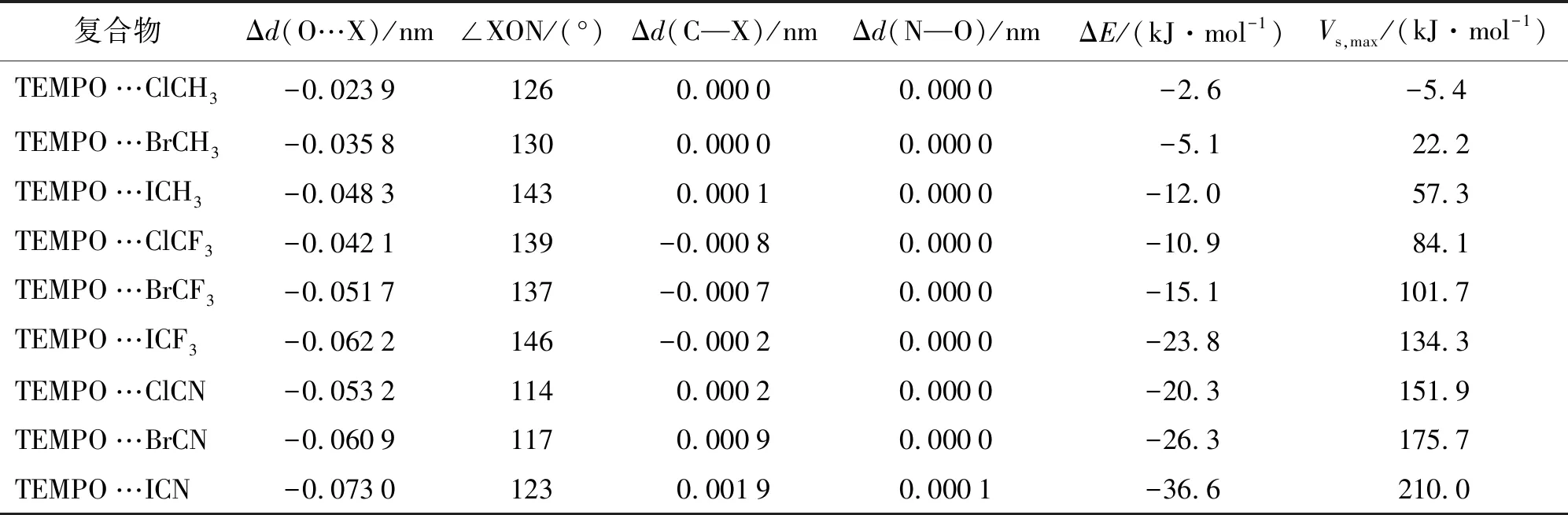
表1 卤键复合物的主要构型数据、分子间结合能及卤化物分子σ-hole处的静电势极大值 Tab.1 Major Configuration Datas of Halogen-bonded Complexes,Binding Energies and Maximum of the Electrostatic Potential at the σ-hole of Halides
2.3 双分子复合物的电子密度拓扑分析
依据Bader提出的分子中原子的量子理论(QTAIM)[24],分子中电子密度分布函数超曲面上的关键点处的电子密度拓扑性质可以表征化学键的强度和性质.图2给出了代表性复合物TEMPO…XY(X=I;Y=CH3,CF3,CN)的分子图.可以看出,TEMPO中的氧原子与XY中的卤原子之间有1个键鞍点(BCP)和连接键鞍点及相应原子的1对键径,表明分子间存在卤键作用.另外,大多数复合物中还存在TEMPO中的氢原子与XY中的卤原子之间的H…X键鞍点,表明复合物中存在二级氢键作用,其增强了分子间相互作用的强度.表2列出了复合物中相应键鞍点处的电子密度拓扑数据:电子密度(ρb)、拉普拉斯量(∇2ρb)、动能密度(Gb)、势能密度(Vb)、能量密度(Hb=Gb+Vb)以及动能密度与势能密度的比值(-Gb/Vb).O…X键鞍点处的ρb和∇2ρb分别为0.008 8~0.021 7 a.u.和 0.039 4~0.081 1 a.u.,数值大小与卤原子(Cl,Br,I)的电负性和取代基(CH3,CF3,CN)的吸电子能力有关.根据电子密度拓扑性质判据[26-27],卤键键鞍点处的∇2ρb> 0,Hb>0,-Gb/Vb>1,说明TEMPO…XY自由基卤键是以静电作用为主的闭壳层非共价相互作用.O…X键鞍点处ρb,Gb,Vb与ΔE间具有良好的线性关系,图3给出了O…X键鞍点处Gb,Vb与ΔE的线性关系图,相关系数分别为0.959 7和0.987 0.卤键键鞍点处ρb,Gb及Vb均可作为衡量自由基卤键强度的指标.
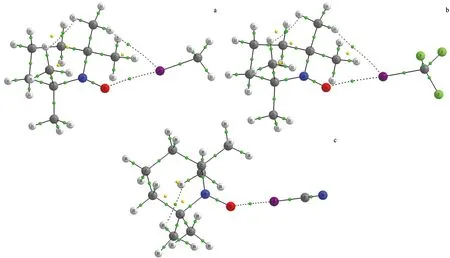
a.TEMPO…ICH3; b.TEMPO…ICF3; c.TEMPO…ICN.图2 卤键复合物的分子图Fig.2 Molecular Graphs of the Halogen-bonded Complexes
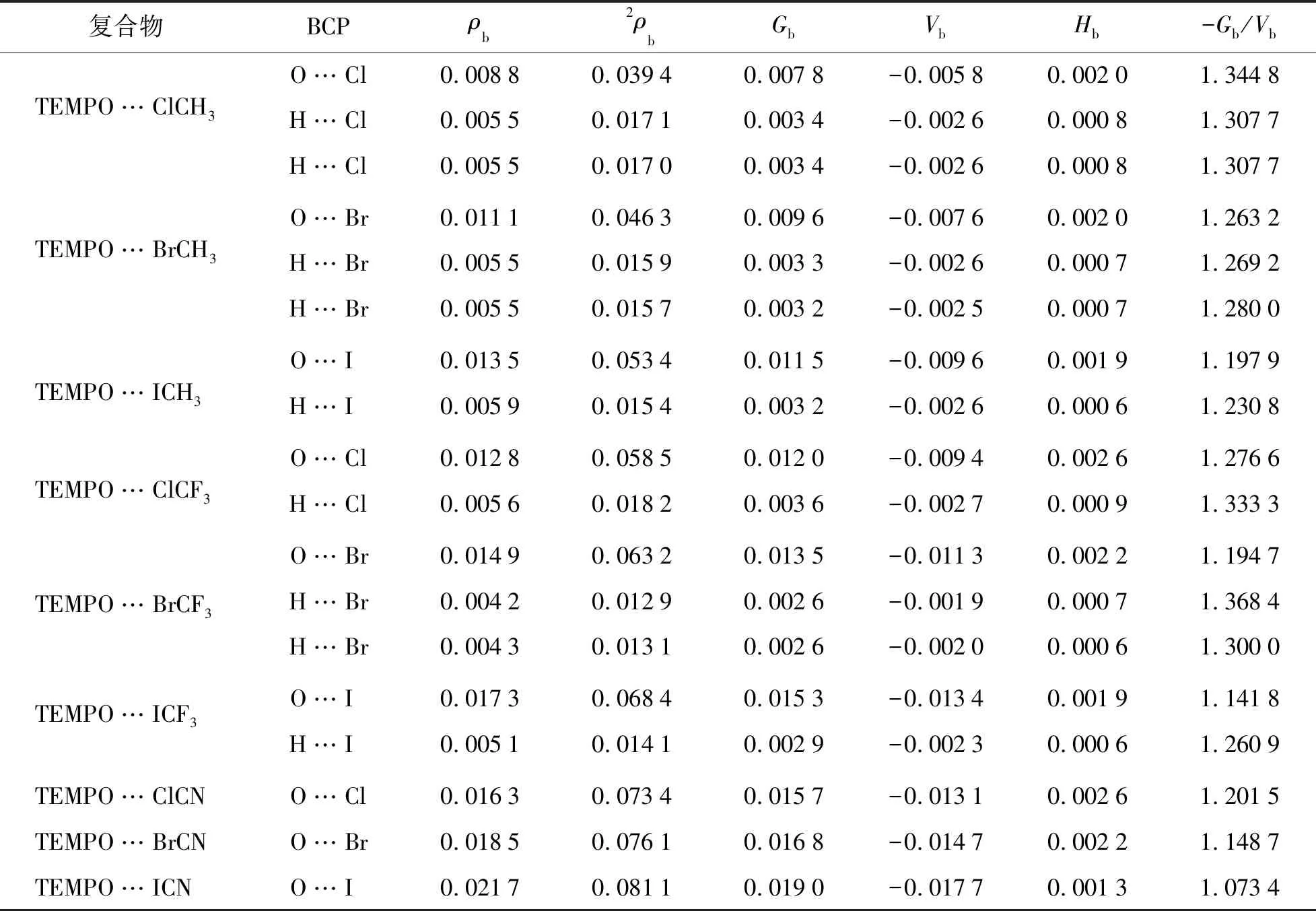
表2 卤键复合物中分子间键鞍点处的电子密度拓扑性质Tab.2 Topological Properties of Electron Density at the BCPs for the Halogen-bonded Complexes a.u.
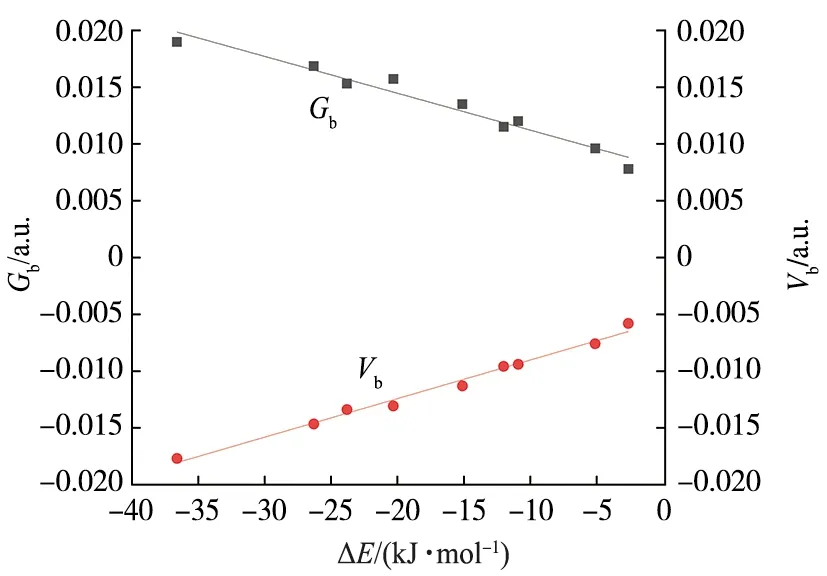
图3 O…X键鞍点处Gb,Vb与ΔE的关系Fig.3 Relationships Between ΔE and Gb,Vb at the O…X BCPs
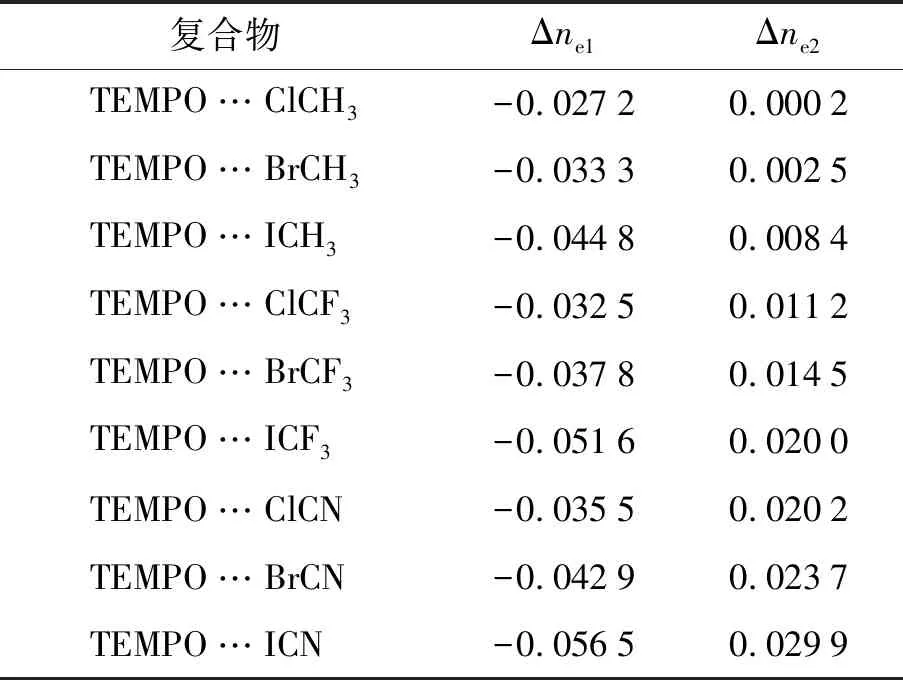
表3 分子间作用区域的积分电荷值Tab.3 Integral Charges in the Intermolecular Regions e
2.4 双分子复合物的分子生成密度差分析
分子生成密度差(MFDD)[28-29]是研究分子间相互作用的重要手段之一,可以直观地表现复合物形成过程中由极化作用引起的分子中电子密度的变化.图4是以卤键复合物所在的XY为平面绘制的二维等值线图(长度单位为bohr).可以看出,极化作用使得分子中的电荷发生了重新排布,TEMPO中氧原子上孤对电子的静电力场导致卤化物分子发生极化,卤原子附近电子密度明显减小(蓝色区域,region1),极化作用使得2分子间相互作用的区域表现为电子密度增加(黄白色区域,region2).从图4中可以明显看出,当卤原子相同时,随着Y基团吸电子能力的加强,分子间极化作用增强,使得蓝色和黄白色区域变得更大.选择region1和region2区域对电子密度变化进行积分,分别得到积分电荷Δne1和Δne2,列于表3中.分析发现,当Y不变时,Δne1和Δne2绝对值随着卤原子X电负性的减小而逐渐增大,当X不变时,Δne1和Δne2绝对值随着Y基团吸电子能力的增强而逐渐增大.
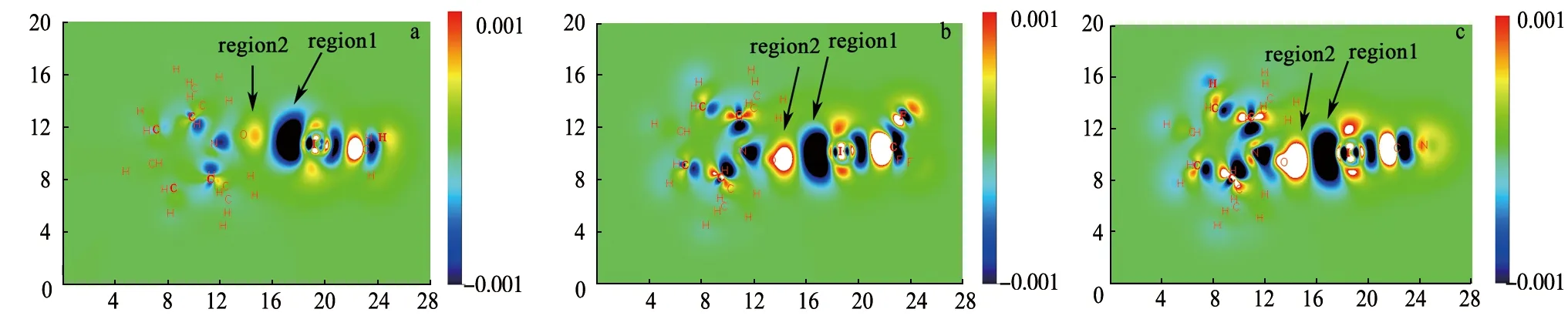
a.TEMPO…ICH3; b.TEMPO…ICF3; c.TEMPO…ICN.图4 复合物的分子生成密度差图Fig.4 The Maps of Molecular Formation Density Difference for the Complexes
2.5 卤键间的协同作用
通过计算双分子复合物TEMPO…XCN(X=Cl,Br,I)的静电势,得到氧原子处Vs,min分别为-111.5,-99.2,-64.6 kJ/mol,比相应单体TEMPO数值更小,可初步判断双分子复合物中氧原子处路易斯碱的给电子能力比相应单体要弱.氮原子处Vs,min分别为-158.7,-163.9,-176.2 kJ/mol,比相应单体XCN数值更大,说明双分子复合物中由于卤键的形成使氮原子具有更强的给电子能力.
将TEMPO与2个XCN(X=Cl,Br,I)构建2种构型的三分子复合物NCX…TEMPO…XCN(Ⅰ)和TEMPO…XCN…XCN(Ⅱ).构型Ⅰ为双分子复合物TEMPO…XCN中的氧原子与XCN中的卤原子作用,构型Ⅱ为双分子复合物TEMPO…XCN中的氮原子与XCN中的卤原子作用.图5是三元复合物NCI…TEMPO…ICN和TEMPO…ICN…ICN的分子图.三分子复合物的能量数据列于表4中,ΔE总表示三分子复合物的总能量,ΔE(A…B)表示双分子复合物中A与B的结合能,ΔE(A…BC)表示在三分子复合物中相对应的A与B的结合能,ΔE′(A…BC)表示三分子复合物与双分子复合物相比结合能的变化,E协表示协同能.
比较表4中结合能数据,ΔE总绝对值随着卤原子电负性的减小依次增强.构型Ⅰ中,ΔE(A…BC)和ΔE(AB…C)的绝对值小于双分子复合物ΔE(A…B)和ΔE(B…C)的绝对值,结合能降低了约20 %,E协均为正值,说明TEMPO…XCN卤键的加入,彼此削弱O…X之间的相互作用,三分子复合物中2个O…X卤键之间存在负的协同作用.E协值随着卤原子电负性的减小而逐渐增大,说明较强的卤键具有较大的协同作用.表4中d(A…B)和d(A…BC)分别表示双分子及三分子复合物中O…X间相互作用距离,可知三分子复合物中d(O…X)大于相应双分子复合物中的数值.三分子复合物构型Ⅰ中,O…X键鞍点处ρb的数值均小于双分子复合物中键鞍点处的电子密度值,表明三分子复合物中的卤键作用减弱.
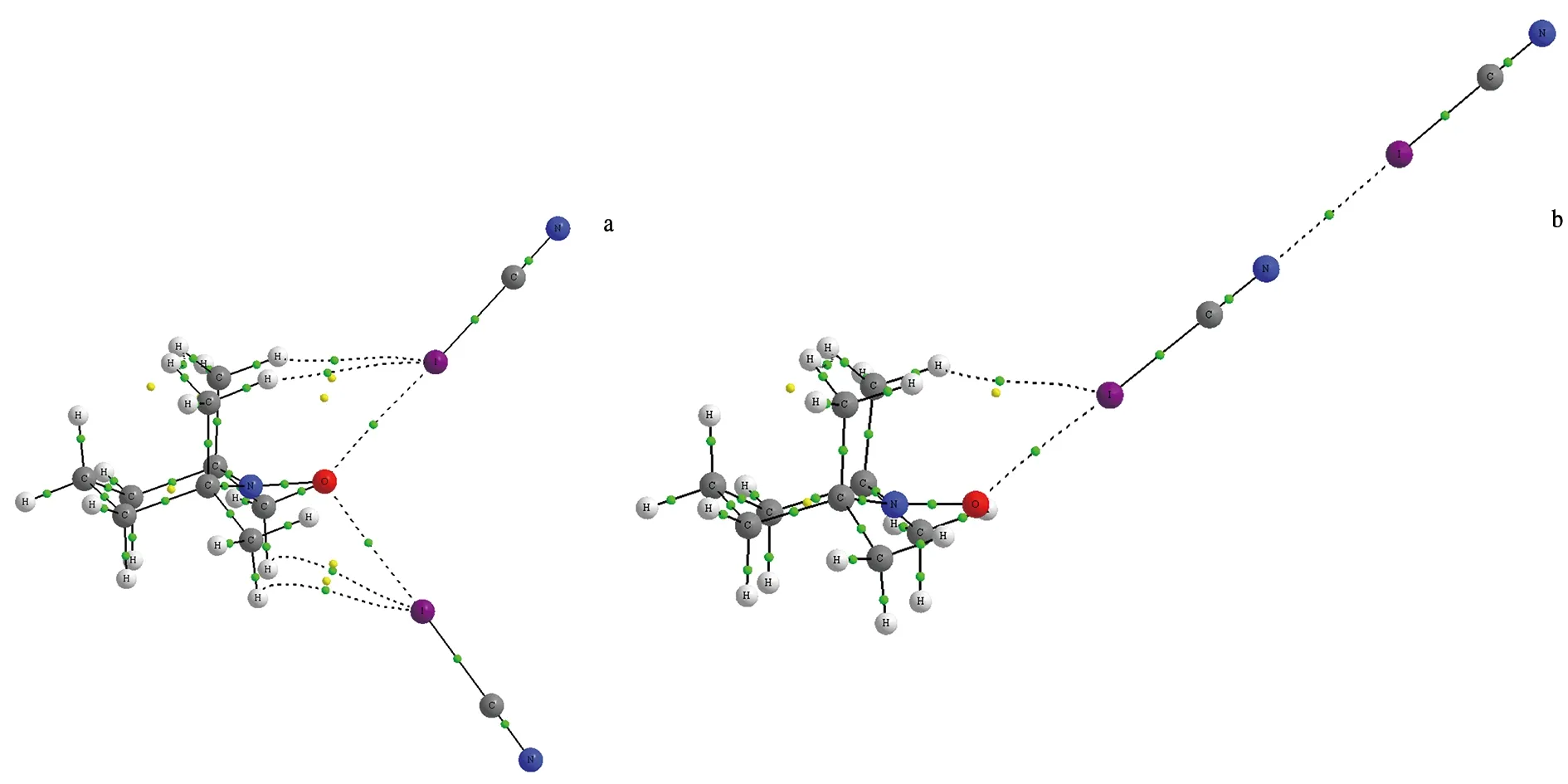
a.NCI…TEMPO…ICN; b.TEMPO…ICN…ICN.图5 三分子复合物的分子图Fig.5 Molecular Graphs of the Trimolecular Complexes
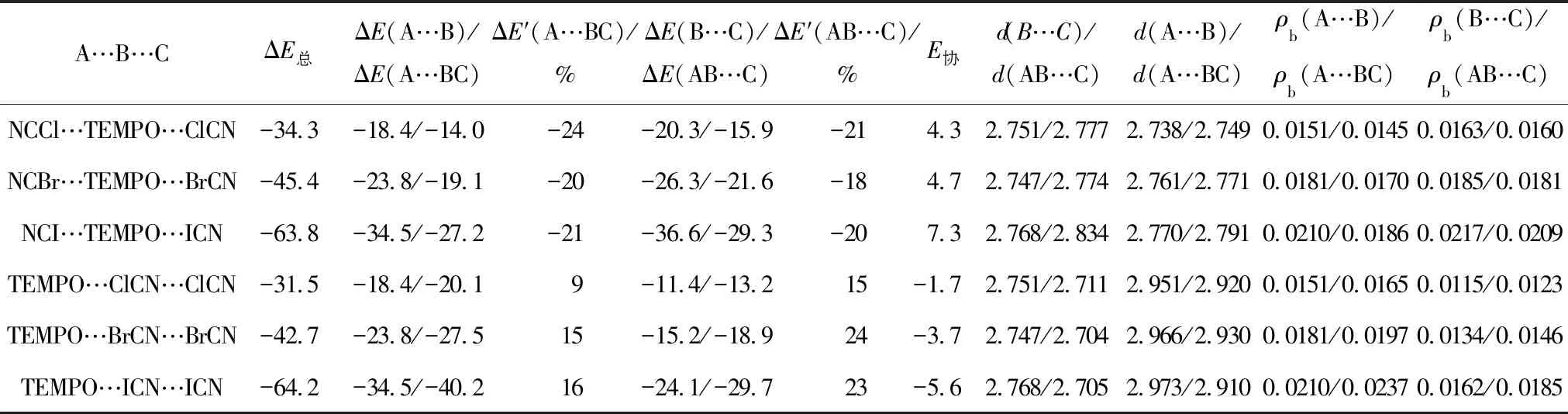
表4 双分子及三分子复合物中O…X间距离、结合能及改变值以及O…X键鞍点处的电子密度Tab.4 The Distances of O…X,Binding Energies and Their Changes,Electron Density at the O…X BCPs in the Bimolecular and Trimolecular Complexes
三分子复合物构型Ⅱ中,ΔE(A…BC)和ΔE(AB…C)的绝对值均大于双分子复合物中ΔE(A…B)和ΔE(B…C)的绝对值,说明XCN…XCN的加入,增强了TEMPO…XCN和XCN…XCN的相互作用.O…X卤键强度大于N…X卤键,因此O…X卤键对N…X卤键的影响(ΔE′(AB…C)为15 % ~ 24 %)大于N…X卤键对O…X卤键的影响(ΔE′(A…BC)为9 % ~ 16 %).协同能E协均为负值,说明复合物中O…X和N…X卤键之间存在正的协同作用.从分子间相互作用距离和卤键键鞍点处的电子密度数值也可以得出相同结论:三分子复合物构型Ⅱ中,相互作用距离d(O…X)和d(N…X)均比相应双分子复合物的作用距离小,O…X和N…X键鞍点处ρb比相应双分子复合物中键鞍点处的ρb大,因此O…X和N…X卤键彼此加强了相互作用强度.
3 结 论
依据量子化学密度泛函理论和分子中原子的量子理论,研究了TEMPO与卤化物XY(X=Cl,Br,I;Y=CH3,CF3,CN)间的双分子及三分子复合物中的自由基卤键,得到卤键复合物的稳定构型、相互作用强度、电子密度拓扑性质,分析了不同构型卤键间的协同效应,主要得出以下结论.
1) 复合物TEMPO…XY(X=Cl,Br,I;Y=CH3,CF3,CN)中分子间结合能为-2.6~-36.6 kJ/mol,卤键强度与卤原子X的电负性以及Y基团的吸电子能力相关,按照X=Cl,Br,I;Y=CH3,CF3,CN顺序依次增强.
2) 双分子及三分子复合物中,自由基卤键是以静电作用为主的闭壳层非共价作用.分子间结合能与卤化物σ-hole处的静电势、O…X键鞍点处电子密度和能量密度、分子生成密度差值间存在良好的线性关系.
3) 在三分子复合物NCX…TEMPO…XCN(X=Cl,Br,I)中,O…X卤键作用弱于其在双分子复合物中的作用,卤键之间存在负协同效应;在TEMPO…XCN…XCN复合物中,O…X卤键和N…X卤键彼此加强,具有正协同效应,较强的卤键对较弱的卤键有更大影响.
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
军民两用技术与产品(2022年1期)2022-06-01
西北工业大学学报(2022年1期)2022-04-22
西华大学学报(自然科学版)(2021年3期)2021-05-17
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
西华大学学报(自然科学版)(2020年6期)2020-10-15
绿色科技(2017年20期)2017-11-10
科技创新导报(2017年19期)2017-09-13
北京航空航天大学学报(2017年10期)2017-04-20
药学研究(2015年11期)2015-12-19
