
卤素取代改良热激活延迟荧光分子CzDBA性能的理论研究
2022-09-20 08:59:42林思榕梁万珍
厦门大学学报(自然科学版) 2022年5期
林思榕,梁万珍
(厦门大学化学化工学院,固体表面物理化学国家重点实验室,福建省理论与计算化学重点实验室,福建 厦门 361005)
近年来,有机发光二极管(OLED)在平板显示器和照明应用领域逐步实现商业化.大量的有机光电材料被广泛研究与开发,其中最受关注的一类是热激活延迟荧光(TADF)材料[1-6].由于受到自旋统计的限制,传统荧光材料的内部量子效率(IQE)的理论极限仅为25%[7-8],但TADF材料可以通过逆向系间窜越(RISC)通道捕获三重态激子,从而有效提高激子利用率.2012年,Uoyama等[9]发现当最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)完全分离时,可以有效降低单-三态能差(ΔEST)从而加速RISC过程.目前,TADF材料的IQE已经能够达到100%[10-11],其电光转换效率,即外量子效率(EQE)也突破了30%[12-13],因此TADF材料被誉为最有前景的第三代光电材料.
TADF材料作为一类重要的电致发光材料常常要求最低单重激发态(S1态)具有明显的电荷转移(CT)特性[14-15],电子通常从给体单元被激发到受体单元上,这就意味着受体单元的吸电子能力十分关键.卤素具有强吸电子能力,且已有研究表明在受体单元上进行卤素取代可以有效缩短延迟荧光寿命,降低三重态激子湮灭带来的损失[16].2013年,Fan等[17]通过对2-苯基苯并噻唑进行卤素取代合成了高效的黄光TADF分子.2018年,Yu等[18]通过在喹喔啉体系中引入F原子得到高效的荧光发射器并对其结构与性质变化进行了深入研究.虽然目前对TADF材料的研究已经取得了不错的进展,但是如何通过合理的分子设计,使TADF材料在发光性能上实现进一步突破仍然面临巨大的挑战.通过理论模拟验证TADF分子的荧光性能,筛选后再进行实验合成,可有效提高TADF分子的研发效率.
2018年,Wu等[13]报道了一种新型TADF分子,即CzDBA(9,10-bis(4-(9H-carbazol-9-yl)-2,6-dimethyl-phenyl)-9,10-diboraanthracene),该分子具有优异的荧光性能,其EQE高达37%.因此,本研究在CzDBA受体环上的不同位点进行卤素取代,设计了12种新型的TADF分子,通过理论计算评估其分子性能(包括HOMO和LUMO的重叠度、CT态特性占比及RISC速率(kRISC)等),分析单-三态能差、重组能、旋-轨耦合等多种因素对TADF材料性能的影响,探讨不同取代位点与不同卤素原子对TADF分子荧光性能的影响.该工作有助于提高对TADF材料工作机理的认识、减少设计成本,并为实验研究提供可行的思路.
1 理论与分析方法
1.1 kRISC
根据费米黄金规则,kRISC的表达式可以写为[19-20]:
(1)

(2)
式中:λ是重组能;kB是玻尔兹曼常数;T是热力学温度,在本研究中均被设定为室温(T=298.15 K).

(3)
式中,i和Q分别代表电子和原子核,ri,Q表示从第i个电子出发到原子核Q之间的矢量算符,pi和si分别表示第i个电子的动量和自旋,结构常数a0=137.037-1,ZQ表示原子核Q的电荷.考虑到三重态的自旋多重度,单-三重态间的旋-轨耦合矩阵元可以按下式计算:
(4)
1.2 长程修正泛函
一般基于传统交换-相关(XC)泛函的TDDFT常常低估CT态的能量,合理描述TADF分子的共轭电子激发需要采用具有足够电子关联效应的理论模型.一些先进的基于波函数的量子化学方法通常能得到与实验较为一致的结果,但随着体系的增大,这些方法的计算成本也会急剧增加[24-26].通过调节范围分离参数(ω)得到的长程修正的XC泛函能够较好地解决这些问题[27].长程修正泛函能准确地预测包括CT激发能在内的激发态性质[28-30],目前已被广泛应用于TADF材料的相关计算.其形式可表示为[31-32]:
(5)
式中:r12是电子间距;erf(ωr12)是误差函数;α和β是常数且0≤α≤1,0≤β≤1,α+β表示在长程极限处所含交换积分的比例.等式右边第一项是DFT的短程部分,第二项是Hartree-Fock的长程交换部分.ω可以通过最小化J2得到[31-32]:
(6)
式中,εHOMO是HOMO的能量,EIP是电离能,N是电子数,c是0或1的实数.精确的Kohn-Sham理论要求HOMO的能量应等于含有N个电子体系的电离能.若忽略电子弛豫的影响,则含有N+1个电子的阴离子体系的电离能近似等于含有N个电子体系的亲和能.
本研究所涉及的激发态相关计算均利用TDDFT/6-31G(d, p)完成,XC泛函采用LC-BLYP*(*表示范围分离参数ω已优化).
1.3 TADF性质分析
HOMO和LUMO的有效分离可以降低ΔEST从而提高逆向跃迁速率,使用两个轨道的重叠度(ξ)来估计空间分离度:
(7)
式中,ΦHOMO表示HOMO的波函数,ΦLUMO表示LUMO的波函数,r表示空间坐标.
均方根误差(RMSD)可用来估计基态与激发态、激发态与激发态间的构型差异程度,定义为:
RMSD=
(8)
式中,x、y、z表示坐标的3个方向,xp与x′p分别表示第p个原子在第1个状态和第2个状态下的x坐标,natom表示体系所含原子总数.
对于电致发光器件,S1态的CT特性占比显得尤为重要.若将给体单元和共轭桥作为片段A并将受体单元作为片段B,则CT态特性占比(ηCT)[33]问题可以简化为每个片段中电子密度和空穴密度的差异:
(9)
式中,ρA,h(r)表示片段A中的空穴密度,ρA,e(r)表示片段A中的电子密度.
1.3节的相关分析均使用Multiwfn3.7程序包[34]得到.
2 结果与讨论
2.1 分子设计与结构优化
如图1所示,CzDBA是一种给体-受体-给体(D-A-D)型TADF分子.本研究以CzDBA分子为初始构型,对其受体单元进行卤素取代,得到12种新型TADF分子.为了区分这些分子并方便后文的叙述,根据取代方式与取代基的不同对这些新分子进行命名,图1中one表示CzDBA受体单元上只有2号位的H原子被取代,x表示CzDBA受体单元上的H原子被“斜位”(2号位和6号位)取代,two表示CzDBA受体单元的2号位与7号位上的H原子被同种卤素原子取代,four表示CzDBA受体单元的2号位、3号位、6号位及7号位的H原子被同种卤素原子取代.例如,fourBr表示CzDBA受体单元的2号位、3号位、6号位及7号位的H原子被Br原子取代后得到的新分子.
使用B3LYP泛函及6-31G(d,p)基组对初始CzDBA分子及本研究设计的12种新型TADF分子进行基态构型优化.通过观察优化后的分子,发现不论是否进行卤素取代,分子两边的给体单元几乎处于同一平面,卤素取代前后分子的整体结构变化不大.由此可以认为该设计是恰当的,并未破坏初始分子的基本构型.
利用在B3LYP/6-31G(d,p)水平下优化后得到的基态波函数通过式(7)计算分子HOMO和LUMO的重叠度(ξ),如图2所示:HOMO主要集中在给体单元,LUMO主要集中在受体单元;12种新型TADF分子的ξ均在10-5量级且均小于初始CzDBA分子(2.08×10-5),其中twoCl与twoBr的ξ最小,均为1.56×10-5.
2.2 ω的优化与ΔEST的计算
为了更加精确地评估激发态性质,使用长程修正泛函LC-BLYP并对范围分离参数(ω)进行优化,基组选择6-31G(d,p).由于ω对体系有较强的依赖性,对所有分子均进行ω的调节,并基于基态结构做垂直激发,分析了S1态与T1态的跃迁占比,结果如表1所示.12种新型TADF分子的ω值范围在0.146~0.162 Bohr-1之间.对S1态而言,不论是CzDBA分子还是本研究设计的12种新型TADF分子,均以HOMO到LUMO的跃迁为主(占比超过50%).由于分子结构中Br原子的强吸电子效应,fourBr分子的S1态的轨道跃迁占比中HOMO到LUMO的跃迁占比更是高达76.22%,比初始CzDBA分子的(60.49%)高了近16个百分点.同一卤素原子取代的TADF分子中,随着卤素原子取代个数的增加,HOMO到LUMO的跃迁占比逐渐增大,如oneF、twoF、xF和fourF分子的HOMO到LUMO的跃迁占比分别为63.20%,65.90%,65.00%和68.48%.对T1态而言,除F取代的4个分子外,其余分子均表现出与S1态相似的轨道跃迁形式.
对TADF材料而言,往往需要S1态具有显著的CT特性,而T1态既可以是CT特性主导也可以是局域激发主导.CzDBA与12种新型TADF分子均为D-A-D型结构,给体单元含有多电子氮,受体单元含有缺电子硼及强吸电子的卤素基团,具有分子内长程CT特性.从表1可以看出:本研究设计的12种新型TADF分子都比初始CzDBA分子(84.96%)拥有更强的S1态CT特性,其中fourCl的S1态CT特性占比最高(89.53%),紧随其后的是fourBr(89.01%).有趣的是,F取代的4个分子的T1态CT特性占比与CzDBA相比都显著降低,均不超过5%;而Cl取代与Br取代后形成的新分子的T1态CT特性占比均在77%以上,较初始CzDBA分子(71.88%)表现出升高的趋势.
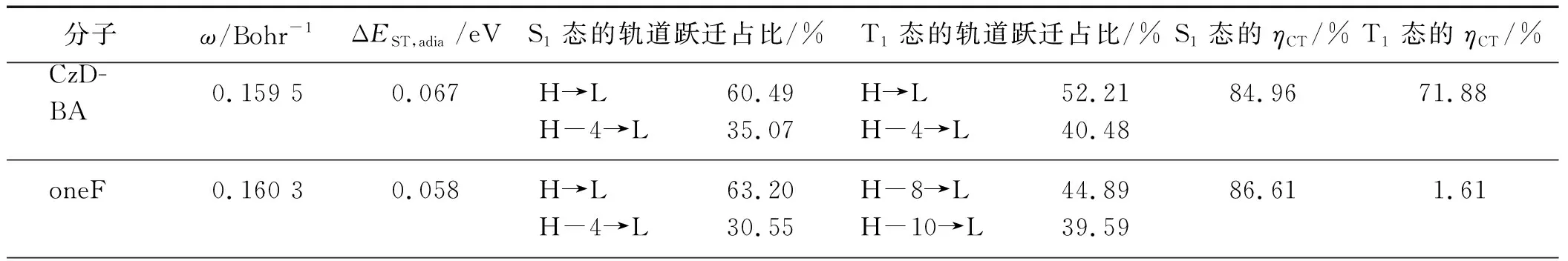
表1 优化的范围分离参数和LC-BLYP*/6-31G(d,p) 水平下计算的单-三态绝热能差(ΔEST,adia)、S1态与T1态的轨道跃迁占比及其CT特性占比

表1(续)
优化激发态结构并通过式(8)对优化后的构型进行比较,结果如表2所示.基态(S0态)与S1态构型、S0态与T1态构型差异最大的均为fourF分子,RMSD分别为0.151 6和0.148 9 nm.S1态与T1态构型变化最大的是twoF,RMSD为0.094 5 nm; oneBr次之,RMSD为0.056 0 nm.
实验测得CzDBA分子的ΔEST为0.033 eV[13],计算得到的ΔEST,adia为0.067 eV(表1).计算结果与实验值在相同量级且较为吻合,这说明本研究选取的泛函是相对成功的.通过理论计算发现,Cl取代和Br取代得到的新分子的ΔEST,adia较初始CzDBA分子均有所减小,其中fourBr的ΔEST,adia最小(0.039 eV),仅为CzDBA分子计算能差的58%.除oneF外twoF、xF和fourF的ΔEST,adia值(0.085,0.094和0.158 eV)均较CzDBA分子有所上升,且随着F原子取代个数的增加能差增幅有明显的增大.
2.3 kRISC
假设式(2)中指数项内的部分为f(λ),则:
(10)
(11)
通常情况下认为降低重组能(λ)有助于加速RISC过程,但经过详细研究Marcus公式(式(1)与式(2)),假设ΔEST为固定值并对方程进行求导后发现:当λ=ΔEST时,Marcus公式中的指数项取得最大值.由
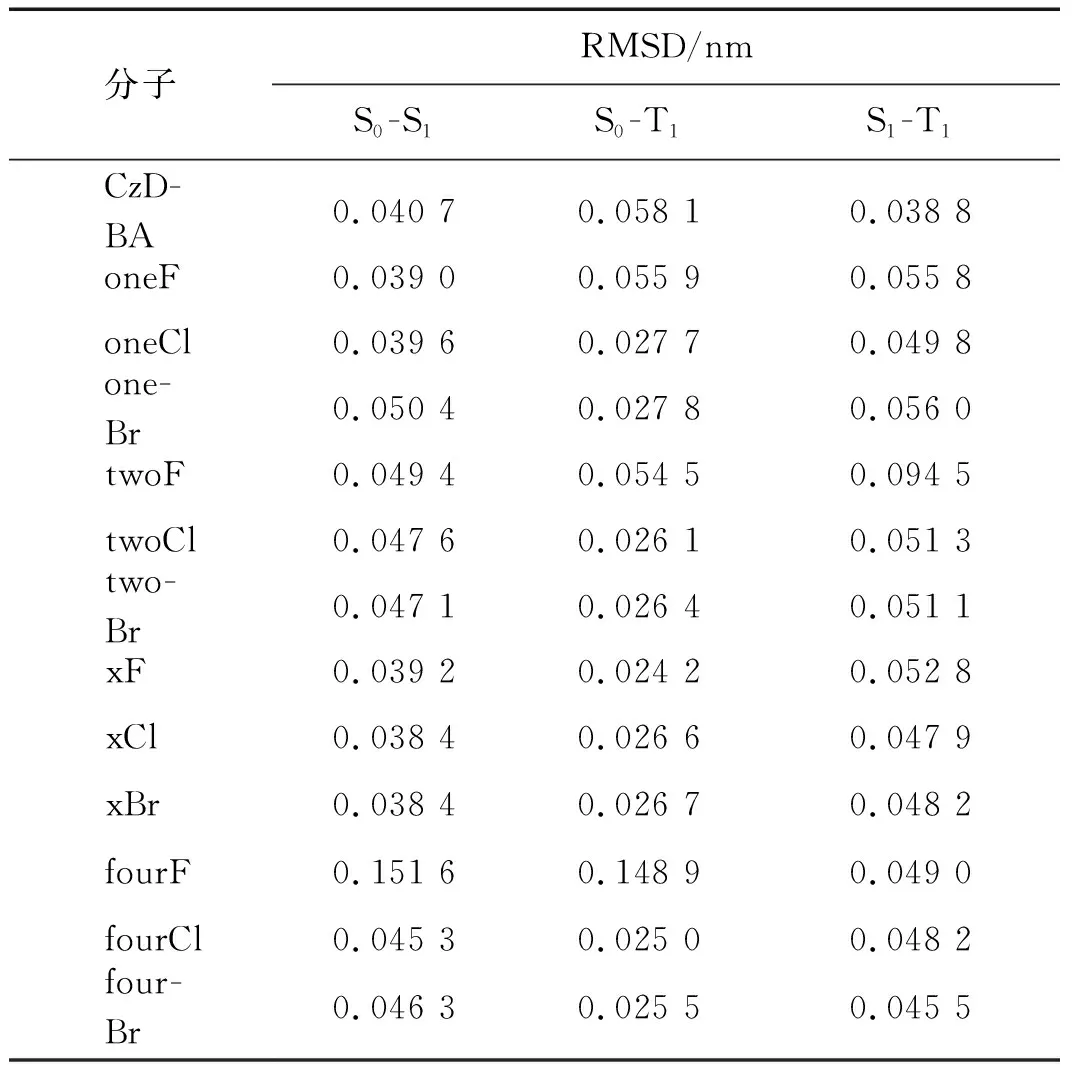
表2 S0态与S1态、S0态与T1态、S1态与T1态之间的RMSDTab.2 RMSD between S0 state and S1 state, S0 state and T1 state, S1 state and T1 state
此可知,只有当λ和ΔEST都很小且数值相近时,才更有利于RISC过程,表3中的结果也证实了这一观点.由于oneCl与oneBr的ΔEST,adia相近,分别为0.065与0.063 eV(表1),对应的ΔEST,adia/λ的值分别为1.032与1.016,其中1.016更加接近于1,则oneBr的指数项数值(0.085)大于oneCl的(0.078).
从表3还可以看出,卤素取代使重组能增大,F取代的4个分子的重组能增幅均较为明显,由于S1态与T1态的构型差异较大,twoF的重组能高达0.295 eV,较CzDBA分子增大约6.6倍.受重组能与ΔEST,adia间平衡关系的影响,虽然本研究所设计的分子的重组能均有所增大,但是其指数项数值并未因此减小.fourCl与fourBr的指数项数值高达0.197与0.212,分别是CzDBA分子的3.3倍与3.6倍,这将使逆向跃迁速率成倍增长,保证了分子的TADF性能.
旋-轨耦合对kRISC有重要的影响,从式(1)可知kRISC与旋-轨耦合值的平方成正比.因此,除ΔEST和重组能外,旋-轨耦合的作用也不容忽视.使用Q-Chem5.2程序包,在LC-BLYP*/6-31G(d,p) 水平下,基于S1态的最优构型进行了旋-轨耦合计算.与掺杂重金属原子的发光材料相比,纯有机分子的旋-轨耦合值一般较小,很少超过1 cm-1.如表3所示,初始CzDBA分子的旋-轨耦合值仅为0.035 cm-1.对应于每种不同的取代方式,旋-轨耦合值均随着卤素原子相对原子质量的增大而增大,这也是重原子效应的一种体现.twoBr拥有最大的旋-轨耦合值(0.118 cm-1),比CzDBA分子增大了2.4倍,紧随其后的是oneBr(0.115 cm-1).在12种新型TADF分子中,one系列与two系列的旋-轨耦合值均较初始CzDBA分子有明显增大.

表3 重组能、重组能与绝热能差之比、指数项数值、旋-轨耦合矩阵元及kRISCTab.3 The calculated reorganization energy,the ratio of reorganization energy to adiabatic energy difference, the value of exponential term,the spin-orbit coupling matrix elements,and the kRISC
利用Marcus公式(式(1)与(2))综合评估了各个可能的因素对RISC过程的影响,并通过计算得到kRISC.如表3所示,F取代的4个分子由于ΔEST与重组能较大,且旋-轨耦合强度也未因F取代而显著提升,所以计算所得的kRISC较初始CzDBA分子有所下降.除xCl外,其余Cl取代分子的kRISC较CzDBA分子均有所上升,Cl取代分子中twoCl的kRISC最大,达到了2.52×105s-1,是CzDBA分子的2.6倍.此外,Br取代的4个TADF分子的kRISC均显著增大,与Cl取代类似,其中twoBr的kRISC(1.78×106s-1)最大,其次是fourBr的(1.77×106s-1),二者的kRISC均达到CzDBA分子的18倍以上,实现了快速的逆向跃迁过程.这表明本研究所设计的分子在理论上具有非常优异的TADF性能.
3 结 论
本研究利用卤素取代CzDBA分子中受体单元的设计理念,在保证最初分子结构框架不变的基础上对其进行修饰改进,所设计的12种新型TADF分子的HOMO-LUMO重叠度较初始CzDBA分子均有所下降,并且S1态具有良好的CT特性.通过研究Marcus公式发现:若ΔEST为定值,并非重组能越小kRISC越大,而是当重组能与ΔEST数值相近时,才能实现最有效的RISC过程.
此外,本研究所设计的12种新型分子在维持基本TADF性能的基础上,显著提升了旋-轨耦合值,其中twoBr和oneBr的旋-轨耦合值分别高达0.118和0.115 cm-1,分别是CzDBA分子的3.4倍和3.3倍,而kRISC与旋-轨耦合值的平方成正比,这将对kRISC带来数量级的改变.
最后计算了原始的以及卤素取代的CzDBA分子的kRISC,结果表明F取代分子由于ΔEST,adia较大等不足,电子无法通过RISC通道快速回到单重态;Cl取代分子中RISC过程最迅速的是twoCl(2.52×105s-1),其kRISC是CzDBA分子(9.71×104s-1)的2.6倍,这主要是因为twoCl的ΔEST,adia与重组能较小且二者数值接近,使得Marcus公式中指数项部分显著增大,从而提高了kRISC.与F取代和Cl取代相比,Br取代分子表现出更加优异的性能:通过Br取代得到的4个分子(oneBr、twoBr、xBr、fourBr)的kRISC均比初始CzDBA分子的更大,其中twoBr和fourBr的kRISC更是达到了初始CzDBA分子的18倍以上.
该工作为TADF分子设计提供了良好的策略,并从理论上证明了它的可行性,其中所设计的Br取代分子在实际应用中具有可憧憬的价值.
猜你喜欢
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08 00:48:08
北京航空航天大学学报(2017年10期)2017-04-20 08:51:23
中学化学(2017年1期)2017-03-17 12:51:55
中学生理科应试(2016年12期)2017-01-07 19:57:10
小学生·多元智能大王(2015年4期)2015-05-18 11:32:05
航天返回与遥感(2014年4期)2014-07-31 17:47:47
无机化学学报(2014年4期)2014-02-28 17:31:08
大学教育(2012年4期)2012-04-29 00:44:03
少年体育训练(2009年3期)2009-04-29 00:44:03
中学生数理化·高一版(2008年12期)2008-06-15 01:31:22
