
水产品中硝基呋喃类药物代谢残留的不确定度评定
2022-09-08 01:31吕燕
浙江农业科学 2022年9期
吕燕
(宁波市农业科学研究院,浙江 宁波 315040)
硝基呋喃类抗生素(nitrofuran antibiotics) 是人工合成的抗感染类药物[1-3],主要是指呋喃唑酮(furazolidone)、呋喃西林(nitrofurazone)、呋喃妥因(furantoin)和呋喃它酮(furaltadone),及其对应的代谢产物有呋喃唑酮(AOZ)、呋喃西林(SEM)、呋喃妥因(AHD)和呋喃它酮(AMOZ)。这类抗生素被广泛用于家禽、家畜、水产、蜂等动物传染病的预防与治疗,但有关研究证明[4-9],硝基呋喃类药物及其代谢物具有较强的毒副作用,可能诱导有机体基因突变,有致畸和诱发癌症的可能,从而被人们高度重视。
如今,我国及大多数欧美国家都禁止使用该类药物,为确保该药物日常检测结果的准确,本文利用《化学分析中不确定度的评估指南》[10]和《测量不确定度评定与表示》[11]等相关技术标准的规定,对用农业部783号公告-1-2006[12]的方法进行前处理并分析检测过程中可能产生的不确定因素,评定最终检测结果的不确定度[9,13-15],以保证最终结果的准确与可靠。
1 材料与方法
1.1 材料
仪器:液相色谱/串联质谱仪、组织捣碎机、分析天平、涡旋振荡器、氮吹仪、容量瓶、离心管、移液枪等。
试剂:甲醇、乙酸乙酯、乙酸铵等。除特殊说明外,其他试剂均为分析纯。实验用水为超纯水。
标准品:AOZ、AMOZ,SEM、AHD,纯度均大于99%。同位素内标:AOZ-D4、AMOZ-D5,SCA-15N2-13C HCL、AHD-13C3,纯度均大于99%。
1.2 方法
1.2.1 标准溶液的配制
混合标准工作溶液的配制:称取AMOZ、AOZ、AHD和SEM标准品各10 mg,用甲醇溶解得到100 mg·L-1的AMOZ、AOZ、AHD和SEM标准储备液。吸取1.0 mL AMOZ、AOZ、AHD和SEM标准储备液用甲醇配制得到1.0 mg·L-1AMOZ、AOZ、AHD和SEM的混合中间标准溶液。再吸取0.1 mL 1.0 mg·L-1AMOZ、AOZ、AHD和SEM的混合中间标准溶液,用水稀释得到0.01 mg·L-1AMOZ、AOZ、AHD和SEM的混合标准工作溶液。内标标准溶液的配制与混合标准溶液的配制方法一致。
1.2.2 样品前处理方法
按照农业部783号公告-1-2006《水产品中硝基呋喃类药物代谢物残留量的测定液相色谱-串联质谱法》的样品处理方式进行提取。
1.2.3 液相色谱-串联质谱条件
色谱柱为C18,液相色谱流速及梯度洗脱程序见表1。采用正离子扫描方式,多反应监测模式(MRM),质谱定性定量离子对信息见表2。

表1 液相色谱梯度洗脱条件

表2 目标化合物液质测定条件
2 结果与分析
2.1 数学模型的建立
根据农业部783号公告-1-2006测定方法及结果计算公式,建立样品中硝基呋喃类代谢物残留量测定的数学模型如下:
X=RcV/Rsm。
(1)
式(1)中:X为检测样品中硝基呋喃类代谢物的含量,μg·kg-1;R为提取的样品待测液中的目标物与内标峰面积比;c为混合标准溶液中硝基呋喃类代谢物的质量浓度,ng·mL-1;V为提取的样品待测液最终的定容体积,mL;Rs为标准液中的目标物与内标峰面积比;m为检测样品的质量,g。
以上各种因素都会对测量结果的不确定度产生影响,其最终合成相对标准不确定度计算公式为:
(2)
式(2)中:urel(c)为合成相对标准不确定度;urel(c0)为标准曲线拟合引入的相对标准不确定度;urel(cs)为标准溶液配制的相对标准不确定度;urel(m)为样品称量的相对标准不确定度;urel(V)为样品前处理过程引入的相对标准不确定度;urel(frep)为重复测量的相对标准不确定度;urel(R)为加标回收率的相对标准不确定度。
2.2 不确定度来源分析
从测定过程和数学模型分析,硝基呋喃类残留测定的不确定度来源如图1所示。

图1 不确定度来源的因果关系
本次测定的不确定度主要来源于以下几个方面:测量重复性引起的不确定度;曲线校准引入的相对标准不确定度;样品前处理过程引入的不确定度;标准物质配制过程中的不确定度;方法回收率引入的不确定度。
2.3 相对标准不确定度分量的评定
2.3.1 标准曲线拟合引入的相对标准不确定度urel(c0)
本实验对4种硝基呋喃类代谢物,选取与硝基呋喃类代谢物相应的同位素内标浓度比,制备了不同浓度点的标准工作液,每个浓度点校准溶液平行测定3次,以标准溶液浓度Ci为横坐标,以标准溶液峰面积比Ai为纵坐标,采用最小二乘法拟合,获得线性回归方程(Y=a+bX)以及线性相关系数r,如表3所示。

表3 硝基呋喃类代谢物回归方程测量结果
线性回归标准曲线拟合引入的不确定度公式如下:
(3)

标准曲线拟合引入的相对标准不确定度urel(c0)结果见式(4)。
(4)
相对不确定度结果列于表4。

表4 相对不确定度
2.3.2 标准溶液配制过程引入的相对标准不确定度urel(cs)
标准溶液配制过程引入的相对标准不确定度的数学模型为:
(5)
标准物质纯度引入的相对标准不确定度见表5。
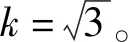
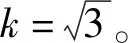
表中:

(6)
根据以上计算的各分量及公式(5)可以计算出标准溶液配制过程中的相对标准不确定度urel(cs):
2.3.3 样品称量过程中引入的相对标准不确定度urel(m)
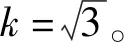
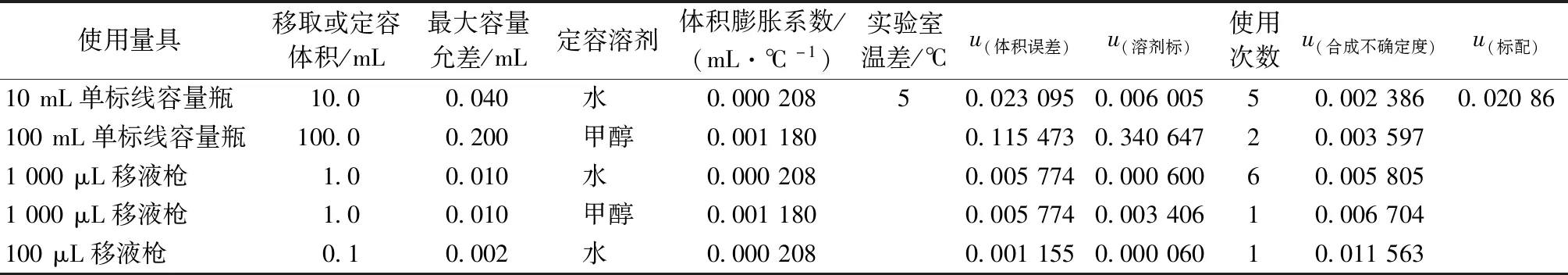
表6 标准曲线配制引入的不确定度
(7)
2.3.4 样品前处理过程中引入的不确定度urel(V)

(8)
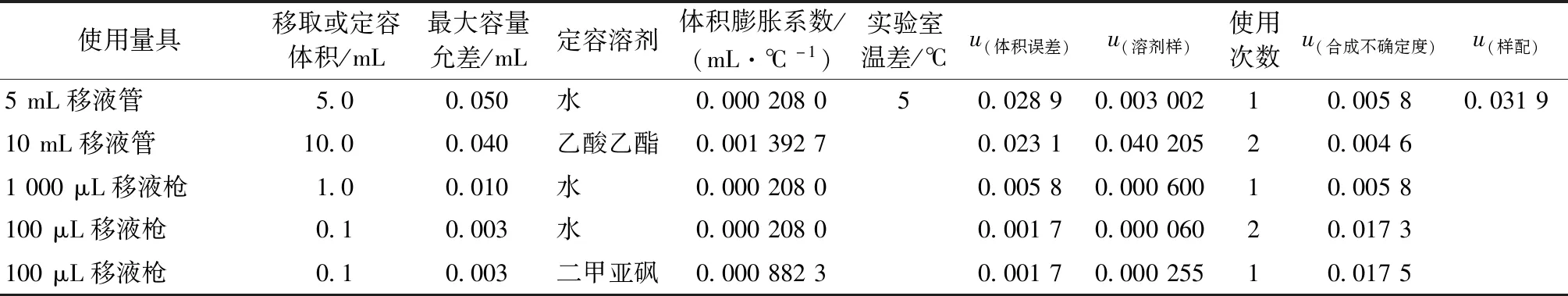
表7 样品制备过程引入的不确定度
2.3.5 整个实验重复性引起的相对标准不确定度urel(frep)
整个实验包括样品称量操作的重复性和进样体积的重复性等。因此,将这些重复性分量合并为总试验的一个分量,利用方法确认的数值将其量化是合理的。称量的重复性属A类标准不确定度,以算数平均值的标准偏差表示。按照1.2节方法进行处理和上机测定,平行测定样品6次,用贝塞尔公式(9)进行计算,结果见表8。公式如下:
(9)


表8 重复测定结果及其相对标准不确定度(n=6)
2.3.6 加标回收率引入的相对标准不确定度urel(R)

(10)

表9 加标回收率结果和加标实验引入的相对标准不确定度
2.3.7 合成相对标准不确定度urel(c)及不确定度uc(X)
以上各项不确定度分量相对独立,不考虑分量间的相关性,将上述各值代入公式(2)中,得出合成相对标准不确定度urel(c),再利用公式(11)来计算不确定度uc(X),最终检测结果取包含因子结果合成不确定度,结果如表10所示。
uc(X)=urel(c)·X。
(11)

表10 合成相对标准不确定度和不确定度
2.4 扩展不确定度
取包含因子k=2(近似95%置信概率),扩展不确定度U=kUc(X),则结果可报告为:X(AOZ)=1.161 6 μg·kg-1,U=0.150 μg·kg-1,k=2;X(AMOZ)=0.858 8 μg·kg-1,U=0.182 μg·kg-1,k=2;X(SEM)=1.027 5 μg·kg-1,U=0.200 μg·kg-1,k=2;X(AHD)=0.933 3 μg·kg-1,U=0.102 μg·kg-1,k=2。
3 小结与讨论
本文按照农业部783号公告-1-2006《水产品中硝基呋喃类药物代谢物残留量的测定液相色谱-串联质谱法》对硝基呋喃类药物进行检测,通过建模、评定和计算测量整个实验过程的各个不确定度分量,得出最终扩展不确定度。结果表明,硝基呋喃类代谢物残留量测定的不确定度主要来自于标准溶液和内标溶液的配制及使用、标准曲线的拟合、加标回收率,其次为测量重复性和样品称样量。因此,在日常检测过程中,应尽量选择高精度器皿,并适当增加测定次数,以最大限度降低整个检测过程中可能带来的不确定度影响,提高检测结果的准确性,为正确评价和使用检测数据提供依据。
猜你喜欢
天津建设科技(2022年2期)2022-05-05
动物营养学报(2022年1期)2022-02-20
世界农药(2021年3期)2021-12-09
口腔护理用品工业(2021年4期)2021-11-02
中国食品(2020年9期)2020-05-26
火工品(2019年3期)2019-09-02
中国科技纵横(2019年23期)2019-02-14
分析化学(2017年6期)2017-06-15
海峡科技与产业(2017年1期)2017-03-04
今日农药(2014年7期)2014-09-15
