
铁死亡诱导药物及其抗癌机制研究进展
2022-09-07 02:10王祥龙葛健文张雅洁张进香吴正红
中国药理学与毒理学杂志 2022年6期
季 鹏,王祥龙,葛健文,张雅洁,柳 熠,张进香,吴正红
(1.泰州学院医药与化学化工学院,江苏泰州 225300;2.中国药科大学药学院,江苏南京 211198)
近年来,基于铁死亡的癌症新疗法受到广泛关注。铁死亡是一种铁依赖性、非凋亡形式细胞死亡方式,这种死亡方式不需要多级信号传导,还可直接损伤细胞膜和遗传物质,过程迅速且效力强大[1]。同时,铁死亡也不会激活DNA损伤应答机制,不易产生耐药性。铁死亡主要是通过抑制谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)或胱氨酸/谷氨酸反向转运体(cystine/glutamate anti⁃porter,System Xc-)活性,减少胞内还原物质如谷胱甘肽(glutathione,GSH)的生成,进而打破细胞氧化还原平衡,从而诱发细胞膜脂质过氧化及膜穿孔,致使癌细胞死亡。目前,针对该机制已研发了多种小分子铁死亡诱导剂,其中RAS选择性致死分子3(RAS selective lethal 3,RSL3),ML210和FIN56可结合或消耗GPX4,而爱拉斯汀(erastin)和柳氮磺胺吡啶(sulfasalazine,SAS)等可通过抑制Sys⁃tem Xc-或消耗胞外胱氨酸/半胱氨酸诱导铁死亡[2]。此外,还有一类诱导芬顿(Fenton)反应的药物(如FINO2)能通过调节Fe2+的氧化水平来诱导癌细胞铁死亡。铁死亡药物作为一种机制新颖的抗癌活性药物潜力巨大,但仍有待深入研究。本文对此3类代表性铁死亡药物的抗癌机制进行综述(图1),旨在为临床以铁死亡为基础的癌症治疗应用及制剂开发提供理论依据。
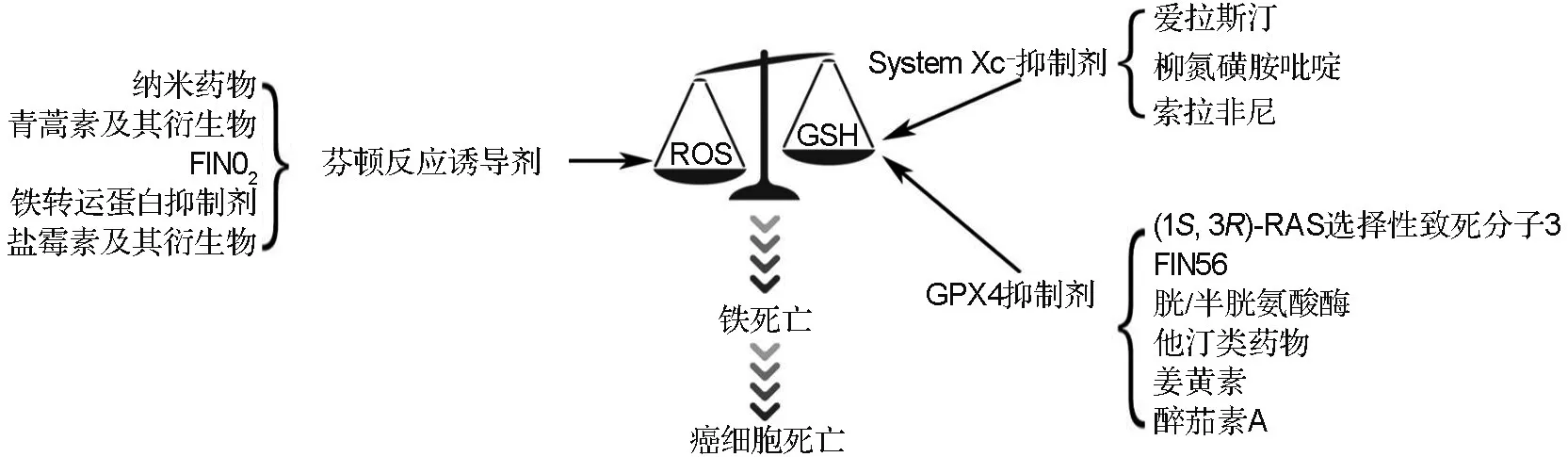
图1 铁死亡诱导药物的抗癌机制及其分类.ROS:活性氧;GSH:谷胱甘肽;System Xc-:胱氨酸/谷氨酸反向转运体;GPX4:谷胱甘肽过氧化物酶4.
1 胱氨酸/谷氨酸反向转运体抑制剂
System Xc-是一种促进胱氨酸/谷氨酸跨膜运输的反转运蛋白,是溶质载体家族成员7A11(solute carrier family 7 member11,SLC7A11)和溶质转运蛋白家族3成员2(solute carrier family 3 member 2,SLC3A2)通过二硫键连接的杂二聚体。System Xc-能介导GSH代谢,促进胱氨酸和谷氨酸跨膜交换,从而使谷氨酸盐被排出细胞,同时胱氨酸在胞内被还原为半胱氨酸[3]。此外,System Xc-还能调节细胞外L-胱氨酸和细胞内L-谷氨酸在细胞膜处的交换,在维持细胞内GSH和细胞外胱氨酸/半胱氨酸氧化还原反应平衡方面起重要作用。研究表明,抑制System Xc-可触发铁死亡。当System Xc-被抑制时,作为GSH生物合成基础的细胞内半胱氨酸生成量减少,间接导致GSH生物合成减少,GSH是GPX4的底物,其合成减少会阻碍GPX4对脂质过氧化物的分解,间接导致脂质过氧化物胞内蓄积,最终促使铁死亡发生。以下就一些典型的System Xc-抑制剂及其抗癌应用进行综述。
1.1 爱拉斯汀
爱拉斯汀是一种已知的胱氨酸/谷氨酸反转运体抑制剂,对其反应机制深入研究时发现了铁死亡现象[4]。爱拉斯汀诱导铁死亡机制是直接抑制System Xc-活性,减少GSH合成,降低GPX4反应底物浓度,从而引起活性氧(reactive oxygen spe⁃cies,ROS)蓄积,最终诱导铁死亡[5]。爱拉斯汀对铁死亡诱导具有高特异性,可刺激热休克因子1(heat shock factor-1,HSF1)依赖性热休克蛋白B1(heat shock protein B1,HSPB1)在癌细胞中表达。敲除HSF1和HSPB1可抑制爱拉斯汀诱导铁死亡效应;反之,用HSF1预处理和将HSPB1过量表达则可促进爱拉斯汀诱导铁死亡[6]。目前,临床应用爱拉斯汀治疗神经母细胞瘤[7]。此外,爱拉斯汀还可诱导人纤维肉瘤细胞HT-1080和人肝癌细胞等发生铁死亡,为爱拉斯汀的临床应用提供新思路[8]。
但爱拉斯汀水溶性差且代谢不稳定的特点限制了其体内应用。科研工作者一直在进行新型爱拉斯汀类似物的研究,咪唑酮埃斯汀(imidazole ketone erastin,IKE)是其中比较成功的一种。细胞学实验结果表明,IKE可有效诱导弥漫性大B细胞淋巴瘤铁死亡,其作用机制与爱拉斯汀类似,但水溶性和代谢稳定性均优于爱拉斯汀[9]。
1.2 柳氮磺胺吡啶
SAS属于磺胺类抗菌药。研究表明,SAS也可通过抑制System Xc-诱导铁死亡,可高效抑制淋巴瘤或其他癌细胞增殖。SAS进入机体后水解成磺胺吡啶和5-氨基水杨酸2部分,其中5-氨基水杨酸是产生药效的主成分,起抗菌消炎和免疫抑制作用,而磺胺吡啶抗菌作用不明显。目前,SAS临床主要用于治疗类风湿关节炎和炎症性肠病等慢性炎症性疾病,但Gout等[10]2001年通过体内外研究发现,SAS还是System Xc-的有效抑制剂,能抑制胱氨酸吸收,导致GSH衰减,最终导致某些特定类型癌细胞死亡。
SAS能通过抑制System Xc-对胱氨酸的吸收而触发铁死亡。在体外实验中,SAS可抑制某些耐药肺癌和乳腺癌细胞增殖[11]。与其他药物相比,SAS对人乳腺癌耐药蛋白具有更强的亲和力,可通过减少自身药物运转增强药效[12]。此外,SAS与传统抗癌药物联用,可减少耐药性和增强药效。有研究发现,达克罗宁和SAS联用可抑制头颈部鳞癌和高表达乙醛脱氢酶3A1的胃肿瘤细胞生长,而单用SAS时,这些肿瘤细胞会迅速产生耐药性[13]。综上,SAS是一种有潜力的铁死亡抗癌药物,但也存在一些不足,如对进展性脑胶质瘤患者疗效不尽如人意和会引发严重神经系统不良事件。因为SAS抑制了谷氨酸外流,而谷氨酸是引发癫痫和激发痛觉的神经递质。因此,SAS用于治疗恶性肿瘤的效果仍待进一步讨论[14]。
1.3 索拉非尼
索拉非尼(sorafenib)是一种蛋白激酶抑制剂,通过抑制System Xc-介导胱氨酸进入细胞,从而导致GSH消耗、内质网应激和脂质ROS积累,最终诱导铁死亡。美国食品和药物管理局(Food and Drug Administration,FDA)现已批准其用于治疗晚期肝细胞癌、晚期肾细胞癌和其他实体肿瘤[15]。此外,与其他药物联用可进一步提高索拉非尼诱导铁死亡的能力,如与青蒿琥酯联用具有协同作用。与单用青蒿琥酯或索拉非尼治疗相比,两药联用显著增强脂质过氧化和铁死亡[16]。Liu等[17]发现,索拉非尼联用肿瘤归巢穿膜肽iRGD可增强纳米药物肿瘤靶向性并加速铁死亡,是一种新的肝癌治疗策略。
2 GPX4抑制剂
GPX4是一种含硒半胱氨酸硒蛋白,是细胞内唯一一个用于脂质体过氧化还原的GPX,具有其他GPX所没有的功能,如通过抑制脂质过氧化发生避免铁死亡过程中细胞死亡。GPX4发挥保护细胞成分免受氧化攻击的重要作用[18]。GPX4可通过催化过氧化物发生还原反应转变为无毒性的脂醇来保护细胞,反应中最常见的还原剂是GSH。GPX4利用GSH作为协同底物,使得脂质中的过氧键转变为羟基,失去其过氧化物性能,从而限制了脂质ROS积累。失活的GPX4也可在半胱氨酸和GSH水平正常的情况下诱导铁死亡发生,这对于根除耐药性恶性肿瘤细胞尤为重要。肿瘤细胞对GPX4高度依赖,目前已可达到在体外根除癌细胞、在体内防止肿瘤复发的效果[19]。
2.1 (1S,3R)-RSL3
GPX4也被认为是通过铁死亡途径杀灭耐药癌细胞的重要靶点。RSL3是GPX4抑制剂的代表性药物之一,可直接靶向GPX4,通过直接抑制GPX4活性触发铁死亡,抑制胶质瘤细胞生长[20]。RSL3以其极高的选择性在铁死亡诱导剂中占据了重要地位,其结构中的亲电性氯乙酰胺结构可通过共价连接GPX4活性位点上46位硒代半胱氨酸残基使其烷基化,从而以不可逆方式直接灭活GPX4,发挥强效抑制作用,使过氧化物酶失活。研究也发现,在RSL3的4种非对映体中,只有(1S,3R)-RSL3对BJ细胞(人皮肤成纤维细胞)具有选择性和强致死性。其中(1R,3R)-RSL3具有与(1S,3R)-RSL3相似的致死性,但由于受基团空间结构(即立体化学)影响,其对HRAS细胞的选择性差,无法作为GPX4抑制剂加以研究和应用[21]。
2.2 FIN56
通过抑制GPX4抑制磷脂过氧化物酶活性也可触发铁死亡。Shimada等[22]以一种有效的选择性铁死亡诱导剂CIL56为研究对象,发现其类似物FIN56是一种新型铁死亡诱导剂。在FIN56结构中,肟基是铁死亡诱导作用的关键,而铁死亡效力也受哌啶基团疏水性影响。FIN56可通过2种不同途径诱导铁死亡:一是在抑制GPX4的同时,借助乙酰辅酶A羧化酶促进GPX4降解,发生铁死亡。二是可通过结合并激活角鲨烯合酶降解GPX4,从而降低GPX4蛋白水平。而CIL56消耗GPX4的同时也会消耗辅酶Q10(coenzyme Q10,CoQ10),因此表现出的效价和特异性低于FIN56,最终被淘汰。CoQ10除了是线粒体呼吸链的一员外,还是一种内源性亲脂性抗氧化剂[23]。
2.3 胱/半胱氨酸酶
胱/半胱氨酸酶是一种能有效降解血清中胱氨酸和半胱氨酸的酶,可通过消耗GSH合成底物(如胱氨酸、半胱氨酸)间接抑制GPX4,从而引发铁死亡[24]。Cramer等[25]发现,胱/半胱氨酸酶可使细胞外L-半胱氨酸和L-胱氨酸含量下降,导致GSH合成减少,ROS含量增高,最终导致肿瘤细胞死亡,同时在动物实验中也展示出良好的耐受性和抗癌效应。此外,胱/半胱氨酸酶还可抑制表皮生长因子受体(epidermal growth factor receptor,EGFR)基因突变的非小细胞型肺癌(non small cell lung cancer,NSCLC)和乳腺癌异种移植瘤的生长。利用胱/半胱氨酸酶调节细胞外胱/半胱氨酸水平的策略可为基于铁死亡抗癌治疗提供新治疗机会。
2.4 他汀类药物
他汀类药物(statins)如辛伐他汀和氟伐他汀等是靶向3-羟基-3-甲基戊二酰辅酶A还原酶(3-hydroxy-3-methylglutaryl CoA reductase,HMGCR)的降脂药物,能降低血液胆固醇水平,同时具有一定抗癌作用。机制上,他汀类药物通过抑制甲羟戊酸通路中焦磷酸异戊烯的生成,抑制GPX4等硒蛋白生物合成,从而促进细胞发生铁死亡[26]。其中辛伐他汀能抑制HMGCR表达、抑制甲戊二羟酸途径和减少GPX4表达,从而诱导癌细胞铁死亡[27]。临床试验数据表明,与传统的抗肿瘤药物三苯氧胺相比,氟伐他汀对过度表达HMGCR的乳腺癌细胞有更强的抗增殖作用[26]。此外,他汀类药物还可与其他疗法联用来提高抑瘤效果,如肝癌患者接受利卡汀联合经导管肝动脉化疗栓塞术(transcatheter hepatic arterial chemoemboliza⁃tion,TACE)治疗的效果好于单纯使用TACE,有效延长了患者生存期;多发性骨髓瘤患者接受沙利度胺、地塞米松和洛伐他汀联合治疗,总生存期和无进展生存期均得到延长,同时体外实验也表明,联合用药诱导细胞凋亡率均高于单一用药。
2.5 姜黄素
姜黄素是一种天然多酚类化合物,具有良好抗癌特性。姜黄素可下调GPX4水平,导致脂质过氧化和ROS积累,从而导致铁死亡。Tang等[28]研究发现,姜黄素降低了肿瘤细胞中GPX4水平,通过激活自噬,在NSCLC中诱导铁死亡,抑制肿瘤生长。Li等[29]发现,姜黄素可以通过上调包括血红素加氧酶1在内的多种铁死亡靶基因表达,从而诱导乳腺癌细胞发生铁死亡,进而抑制乳腺癌。因此,铁死亡可能是天然多酚类药物的抗癌作用机制之一。
2.6 醉茄素A
醉茄素A(withaferin A,WA)是FDA批准的一种天然甾体内酯类化疗药物,具有良好治疗应用前景[30]。WA是一种天然铁死亡诱导剂,可通过抑制GPX4的蛋白水平和活性诱导铁死亡。WA能抑制异种神经母细胞瘤细胞的增殖和耐药,在体内神经母细胞瘤中显示良好抗肿瘤活性,从而开辟了神经母细胞瘤治疗研究新领域[31]。
3 芬顿反应诱导剂
3.1 纳米药物
肿瘤细胞内存在芬顿反应,即H2O2在内源性Fe2+/Fe3+催化下生成羟基自由基,导致脂质过氧化物异常蓄积,促进肿瘤细胞铁死亡[32]。然而肿瘤细胞内通常存在H2O2浓度很低或芬顿反应单一疗法疗效不足等问题。纳米材料具有优良的理化性质,将其开发成多功能铁死亡纳米药物,发挥更加强大肿瘤抑制作用是一种新的制剂研究思路[33]。Li等[34]将 H2O2包裹在 Fe3O4-乳酸-葡萄糖酸共聚物〔poly(lactic-co-glycolic acid),PLGA〕纳米载体亲水核中,形成H2O2/Fe3O4-PLGA聚合体,通过Fe3O4释放铁离子,从而加速芬顿反应,产生过量ROS,杀灭子宫颈癌细胞HeLa细胞。Huo等[33]将葡萄糖氧化酶(glucose oxidase,GOD)和Fe3O4纳米粒同时载入大孔径、可生物降解的树状二氧化硅纳米粒(dendritic silica nanoparticle,DMSN)中构建了一种多功能纳米药物GOD-Fe3O4@DMSN,能增加癌细胞内H2O2浓度。一方面,该系统中GOD能通过氧化癌细胞中的葡萄糖产生H2O2,而Fe3O4释放的外源性铁离子也为脂质过氧化物的产生提供H2O2,可增强芬顿反应;另一方面,GOD进入癌细胞后会消耗大量葡萄糖,阻断肿瘤细胞的能量来源,实现“饥饿”疗法,抑制肿瘤细胞增殖。研究发现,该纳米药物生物相容性好,可诱导人恶性胶质瘤细胞U87L铁死亡。Zheng等[35]基于铁离子与单宁酸的配位作用,合成了金属有机框架包裹P53质粒的制剂,此制剂进入癌细胞后可响应胞内微酸性环境,不仅能释放铁离子,催化芬顿反应,产生细胞毒ROS,还能将p53导入癌细胞进行基因治疗,有效抑制了4T1小鼠乳腺瘤生长和转移。Shen等[36]用纳米载体Fe3O4/GD2O3同时负载常用抗癌药物顺铂(cisplatin,Pt)和乳铁蛋白(lactoferrin,LF),并将它们偶联到LF和精氨酸-甘氨酸-天冬氨酸(arginineglycin-aspartic acid,RGD)二聚体上,构建了纳米药物FeGd-HN@Pt@LF/RGD2。该纳米药物中修饰的RGD肽可有效识别癌细胞表面整合素αvβ3受体,促进纳米粒靶向癌细胞,而LF和药物能增加癌细胞中反应物(Fe2+,Fe3+和H2O2)胞内浓度,从而加速芬顿反应,增强诱导铁死亡能力。体内外研究结果表明,该纳米药物可显著抑制人脑星形胶质母细胞瘤U-87MG细胞的增殖和小鼠胶质母细胞瘤的生长。
3.2 青蒿素及其衍生物
青蒿素(artemisinin)及其衍生物主要包括青蒿素、双氢青蒿素(dihydroartemisinin)、青蒿琥酯(artesunate)等,大都具有较好的抗肿瘤活性。青蒿素是从黄花蒿中提取得到的含过氧基团倍半萜内酯,最初开发为抗疟疾药物。有研究发现,其可通过调控铁代谢相关基因表达诱导ROS生成,致使癌细胞铁死亡[37]。双氢青蒿素是一种含过氧桥的倍半萜内酯类化合物,可通过加速细胞内铁蛋白降解,提高细胞内铁离子浓度,增加体内ROS含量,同时增强细胞对铁死亡的敏感性,诱导细胞铁死亡,对多种肿瘤均有抑制作用,具有抗肿瘤功效[38]。青蒿琥酯是青蒿素的水溶性衍生物,是治疗疟疾的一线药物。近年来,研究发现其有激活溶酶体作用,并可通过增加铁蛋白水解提高铁离子浓度,进而通过铁依赖性诱导铁死亡。胱氨酸/谷氨酸转运蛋白(cystine/glutamate transporter,xCT)是铁死亡核心负调节因子,转铁蛋白受体(transferrin receptor,TFRC)mRNA是一种铁死亡正调节因子。Zhang等[39]以NSCLC为研究模型,发现青蒿琥酯可下调xCT,上调TFRC mRNA水平,通过诱导ROS依赖性细胞凋亡或铁死亡高效抑制NSCLC活性。
3.3 FINO2
FINO2是基于铂酸天然产物类似物开发出的一种含超氧化物的1,2-二烷,类似于青蒿素的有机过氧化物,可通过发生芬顿反应氧化Fe2+产生ROS,且能间接失活GPX4,二者联合诱导癌细胞发生铁死亡[40]。由于具有高亲脂性,FINO2可能会蓄积在膜脂双层中,直接氧化多聚不饱和脂肪酸,致使细胞膜穿孔甚至破裂。FINO2与含有过氧键的青蒿素类化合物的差异在于它影响铁死亡的方式是在铁氧化和GPX4活性丧失的基础上实现的,且FINO2诱导细胞死亡不依赖于胱天蛋白酶或线粒体外膜通透性改变,能优先激活铁死亡而非其他形式细胞死亡[41]。综上,基于 FINO2的深入研究,或许可获得一类新铁死亡诱导剂,使其在治疗癌症方面比青蒿素更有潜力。
3.4 铁转运蛋白抑制剂
铁转运蛋白抑制剂主要有西拉美辛(siramesin)和拉帕替尼(lapatinib)等。西拉美辛最初用于治疗抑郁症,对人类有很好耐受性。拉帕替尼是一种有效的EGFR和人类表皮生长因子受体2(human epidermal growth factor receptor 2,ErbB2)双酪氨酸激酶抑制剂,具有抑制ErbB2和EGFR过表达的癌细胞增殖的能力。最近一项研究表明,两药联用可产生协同作用,并诱导人乳腺癌细胞MDAMB-231和SKBR3铁死亡,其机制是通过降低铁转运蛋白和铁蛋白表达,增加转铁蛋白表达,从而增加游离铁含量;而过量的铁催化芬顿反应,产生羟基自由基,并增加ROS产生,导致铁死亡[42]。
3.5 盐霉素及其衍生物
盐霉素(salinomycin)是一种从白色链霉菌菌株中分离出来的单羧基聚醚类广谱抗生素,对革兰阳性菌、真菌和寄生虫效果较好。最近研究发现,盐霉素在体外和体内有抗肿瘤活性,且其合成衍生物铁霉素的抑瘤效力是其10倍[43]。盐霉素和铁霉素抗癌机制是通过激活癌细胞溶酶体内铁反应元件结合蛋白2和转铁蛋白受体,快速降解铁储存蛋白铁蛋白,增加溶酶体中游离铁含量,过量铁催化芬顿反应,并增加ROS产生,导致铁死亡[44-45]。
4 其他机制铁死亡药物
铁死亡被认为仅受GPX4和自由基捕获抗氧化剂控制[46]。由于不同癌细胞系对GPX4抑制剂敏感性差异很大,使GPX4诱导铁死亡治疗癌症应用受限,独立于GPX4的铁死亡信号通路及药物的探索迫在眉睫。最新研究发现,铁死亡抑制蛋白1(ferroptosis suppressor protein 1,FSP1)是有效的铁死亡抵抗因子,在铁死亡中具有重要作用。FSP1与不同细胞膜结合,并通过还原CoQ10阻止脂质过氧化而抑制铁死亡,为诱导铁死亡抗癌相关药物开发提供了重要参考策略[47]。Doll等[46]合成了FSP1抑制剂,可抑制FSP1诱导铁死亡,在多种表达FSP1的癌细胞(如人大细胞肺癌细胞H460、人乳腺癌细胞MDA-MB-231和人纤维肉瘤细胞HT1080)中都显示出抑癌活性。
5 结语
诱导癌细胞发生铁死亡是目前公认的、颇具前景的癌症治疗策略,在抑制肿瘤生长和增殖方面起着至关重要的作用。目前,铁死亡相关研究正处于迅速发展阶段,铁死亡药物有望成为一种极具前景的癌症诊断和治疗干预方案,对抗肿瘤药物的发展也具有重要意义,本文对铁死亡诱导药物(表1)及各类代表药物的抗癌机制进行了系统综述。未来,铁死亡诱导药物及其他小分子铁死亡药物有潜力成为抗癌用药主力军。然而,现阶段开发的部分铁死亡药物存在生物利用度较差和靶向性不足等问题,阻遏了其临床前景。如在目前研究的基础上将小分子药物组装特异受体结合癌细胞靶点进行靶向治疗,将会极大程度提高该类药物在癌症治疗中的地位。最近涌现出的将促铁死亡药物与传统免疫治疗或放射治疗相结合的疗法展现出了显著的肿瘤抑制效果,这不仅促进了肿瘤传统疗法的进步,也让研究者们对铁死亡治疗肿瘤的潜力有了更高期待。

表1 铁死亡诱导药物及其抗癌应用
猜你喜欢
台港文学选刊(2022年2期)2022-04-14
疯狂英语·新阅版(2021年9期)2021-10-30
小学生学习指导(低年级)(2021年6期)2021-07-19
小学生学习指导(中年级)(2021年6期)2021-06-19
小学生学习指导(高年级)(2021年6期)2021-06-19
昆明医科大学学报(2021年1期)2021-02-07
中华养生保健(2020年4期)2020-11-16
祝您健康(2020年5期)2020-05-14
好日子(2018年9期)2018-10-12
中国医药导报(2011年27期)2011-12-31
