
炎性小体在诱导脊髓损伤神经炎症中的作用
2022-09-07 01:24周蕊寒刘建成张安仁
中国生物化学与分子生物学报 2022年4期
周蕊寒, 刘建成, 张安仁,3)*
(1)成都中医药大学养生康复学院,成都 610075;2)中国人民解放军西部战区总医院康复医学科,成都 610083;3)同济大学医学院附属上海第四人民医院康复医学科,上海 200080)
脊髓损伤(spinal cord injury, SCI)是一种具有高致残率的创伤性疾病,以损伤平面以下的神经功能障碍为主要表现[1]。SCI治疗与康复一直是医学领域的重大难题。现代医学显著提高了患者的存活率,但在改善损伤的神经功能方面进展甚微。脊髓损伤分为原发性损伤和继发性损伤。其中,继发性损伤是造成脊髓损伤后神经功能障碍的主要原因,而炎症反应是继发性损伤阶段最重要病理过程,急性期通过抑制神经炎症来减轻继发性损伤被认为可减轻神经功能损害而达到神经保护作用[2]。近年的研究发现,炎性小体(inflammasome)参与引发脊髓损伤后的神经炎症。炎性小体是由胞浆内模式识别受体(pattern recognition receptors, PRRs)参与组装的一类蛋白质复合体。作为介导固有免疫反应的重要蛋白质,在感染、损伤和自身免疫性疾病时,能充当感染或损伤刺激的传感器,快速识别病原相关分子模式(pathogen associated molecular patterns, PAMPs)或损伤相关分子模式(damage associated molecular patterns, DAMPs),并激活下游的胱天蛋白酶1(cysteinyl aspartate specific proteinase-1, Caspase-1)[3]。胱天蛋白酶1可以裂解促炎细胞因子白介素1β(interleukin 1β, IL-1β)和白介素18(interleukin 18, IL-18)的前体,使其转化为成熟的IL-1β和IL-18,同时切割孔形成蛋白Gasdermin D(GSDMD),诱导一种以细胞肿胀破裂并释放细胞内容物为特征的程序性细胞死亡方式细胞焦亡(pyroptosis)[4]。促炎症细胞因子和细胞焦亡释放的胞内物质都可作为促炎信号引发炎症反应。在脊髓损伤后,脊髓神经元和胶质细胞中的炎性小体相关蛋白质表达增加,导致IL-1β、IL-18释放和神经细胞焦亡,加重继发性炎症反应。靶向抑制炎性小体的激活会减轻炎症反应,促进神经细胞存活,达到神经保护作用。因此,炎性小体有望成为脊髓损伤治疗的新靶点。本文拟对炎性小体的结构及其在脊髓损伤中的作用、激活机制和治疗前景进行综述,以期为后续研究提供思路。
1 炎性小体的结构
炎性小体(inflammasome)本质上是由多种蛋白质组装的蛋白质复合物,大多由受体蛋白质、衔接蛋白质、效应蛋白质3个部分组成。受体蛋白质主要是指来源于Nod样受体(nod-like receptors, NLRs)家族与PHYIN(pyrin and HIN domain)家族的模式识别受体。目前,研究较多的炎性小体结构(Fig.1)主要由NLRs家族的NLRP1、NLRP3、NLRC4(IPAF)和PHYIN家族的AIM2(absent in melanoma 2)等受体蛋白质作为主要框架组装[5]。NLRP1、NLRP3、NLRC4(IPAF)蛋白质中间都有1个核苷酸结合和寡聚结构域(nucleotide-binding and oligomerization domain),又称NACHT结构域,可调节自身寡聚和炎性小体的组装;C-端为富含亮氨酸的重复序列结构域(leucine-rich repeats, LRR),在感应配体和自动调节中发挥作用;N-端为胱天蛋白酶募集域(caspase recruitment, CARD)或吡啶域(pyrin domain, PYD),介导下游的信号传导;AIM2蛋白质是一类DNA感受器,C-端为HIN200结构域,识别并结合自体或异体的DNA,N端为PYD结构域。凋亡相关斑点样蛋白(apoptosis-associated speck-like protein, ASC)作为衔接蛋白质,同时具有PYD结构域和CARD结构域。胱天蛋白酶1的前体(pro-caspase-1)为效应蛋白质,具有CARD结构域。当受体蛋白质被激动剂激活后,会随之吸引ASC和胱天蛋白酶1的前体组装成一个多蛋白质复合的炎性小体,复合体中胱天蛋白酶1的前体自动水解释放P20和P10亚基,形成有活性的胱天蛋白酶1。活化的胱天蛋白酶1一方面对pro-IL-1β、pro-IL-18的Asp和Ala位点进行水解,使其转化为有活性的IL-1β、IL-18[6];另一方面切割孔形成蛋白GSDMD,产生GSDMD-N和GSDMD-C两个肽段,其中GSDMD-N可以结合膜脂质,破坏其完整性,使胞膜破裂引起焦亡[7-8]。在形态学上,焦亡兼有坏死(necrosis)和凋亡(apoptosis)的一部分特征,但又不同于坏死和凋亡。主要表现为细胞膜结构的完整性受到破坏,形成非选择性的孔洞,细胞肿胀破裂和释放胞内物质;同时也表现出细胞核浓缩和染色质DNA断裂等凋亡的特征,最后引起细胞的渗透性溶解[9]。

Fig.1 The structure of common inflammasomes Domains: CARD, caspase recruitment domain; FIIND, domain with function to find; LRR, leucine-rich repeat; NACHT, nucleotide-binding and oligomerization domain; PYD, pyrin domain; HIN-200, HIN-200 DNA-binding domain.Removal of the CARD domain of caspase-1 by autocleavage at the indicated sites results in the formation of the active caspase-1 p10/p20 tetramer
NLRP3是研究最多的炎性小体,具有典型的3类蛋白质结构;NLRP1炎性小体是研究最早的炎性小体,也是结构最为复杂的炎性小体之一。这2种炎性小体是脊髓损伤中最常见的炎性小体。NLRP1含有独特的功能查找结构域(domain with function to find, FIIND),其通过自身蛋白质水解参与炎性小体激活[10]。人类NLPR1蛋白同时具有PYD和CARD结构域,因此,ASC对人类NLRP1炎性小体的激活并非必不可少,但是ASC可以增强NLRP1对胱天蛋白酶1的激活[11];de Rivero Vaccari等[12]在大鼠脊髓损伤模型中也得到了类似的结果。此外,在中枢神经系统中的NLRP1炎性小体与外周巨噬细胞中的NLRP1炎性小体不同。首先,中枢神经系统中的NLRP1炎性小体在活化前以部分预组装的状态存在[12-13]。即在NLRP1炎性小体各成分之间存在蛋白质-蛋白质相互作用,在受到应激时可促进其成分蛋白质进一步结合,这种状态有助于免疫反应在损伤后快速激活。其次,在结构上,除了NLRP1、胱天蛋白酶1、ASC蛋白以外,还包括X连锁的凋亡抑制蛋白(X-linked inhibitor of apoptosis protein, XIAP)。XIAP是一种凋亡抑制因子,可直接抑制胱天蛋白酶。当XIAP裂解成小片段,其抑制胱天蛋白酶的能力降低[14]。因此,脊髓损伤诱导的XIAP裂解可降低胱天蛋白酶1激活的阈值。
2 炎性小体与脊髓损伤
2.1 炎性小体在脊髓损伤后的作用
在脊髓原发损伤的瞬间,外力直接损伤脊髓组织,神经细胞膜破裂,从而导致不可逆的神经细胞坏死。在继发损伤阶段,细胞坏死诱导的细胞内物质例如谷氨酸、三磷酸腺苷(ATP)、活性氧(reactive oxygen species, ROS)和组织蛋白酶B(cathepsin B, CTSB)等[15]快速释放,这些物质作为DAMPs可被PRRs识别,并组装激活炎性小体,启动固有免疫应答。固有免疫反应在脊髓损伤的病理中发挥至关重要的作用。小胶质细胞是中枢神经系统中主要的固有免疫细胞,可通过PRRs来感知组织损伤和外来感染[16]。损伤发生后,小胶质细胞最先在损伤部位募集,并在细胞形态上发生极化,其M1表型释放的促炎细胞因子、趋化因子和ROS等促进外周固有免疫细胞(巨噬细胞和中性粒细胞等)和适应性免疫细胞(T细胞和B细胞)募集到损伤部位,以及进一步激活附近的神经细胞,引发炎症级联反应[17]。NLRP3炎性小体主要在小胶质细胞中组装[18-19],通过激活胱天蛋白酶1,介导小胶质细胞释放促炎细胞因子IL-1β和IL-18。IL-1β是炎症的关键引发剂,对细胞活化和细胞因子的产生都具有重要贡献[20];IL-18也被认为是T细胞和自然杀伤细胞中干扰素-γ的重要调节剂[21],都能促进适应性免疫应答激活。一般认为神经元不充当抗原提呈细胞,免疫功能有限,在神经元中组装激活的NLRP1[12]和NLRP3[22]炎性小体通过介导IL-1β、IL-18成熟释放,为神经元提供了参与固有免疫的能力。焦亡也是脊髓损伤后神经细胞死亡的重要方式,细胞破裂时释放的胞内物质作为促炎信号也参与引发炎症反应。Li等[23]发现,在脊髓损伤后神经元的焦亡是在凋亡后出现,凋亡在损伤后第1 d达到峰值,而焦亡的峰值出现在损伤后第3 d,并且与凋亡相比,焦亡的持续时间更久。
总之,炎性小体作为损伤刺激的传感器,在脊髓损伤后的小胶质细胞和神经元中迅速启动固有免疫应答。激活下游胱天蛋白酶1介导的细胞焦亡途径,释放促炎细胞因子IL-1β、IL-18,导致适应性免疫应答激活,扩大炎症级联反应。
2.2 炎性小体的激活机制
炎性小体激活具有一个复杂的机制。以目前研究较多的NLPR3炎性小体来具体讨论,NLRP3炎性小体的激活通常需要2个步骤:1)初始启动信号增加炎性小体和促炎因子的前体转录并触发翻译后修饰,该信号由Toll样受体4(toll-like receptor 4, TLR4)-核因子κB(nuclear factor кB, NF-κB)途径诱导,即Toll样受体受到刺激后通过调节NF-κB转录因子增加NLRP3和pro-IL-1β、pro-IL-18的转录[24];2)触发炎性小体组装激活。目前研究认为,触发NLRP3炎性小体组装激活的因素涉及K+外流、线粒体Ca2+超载、ROS生成和溶酶体破裂等生物事件(见Fig.2)。
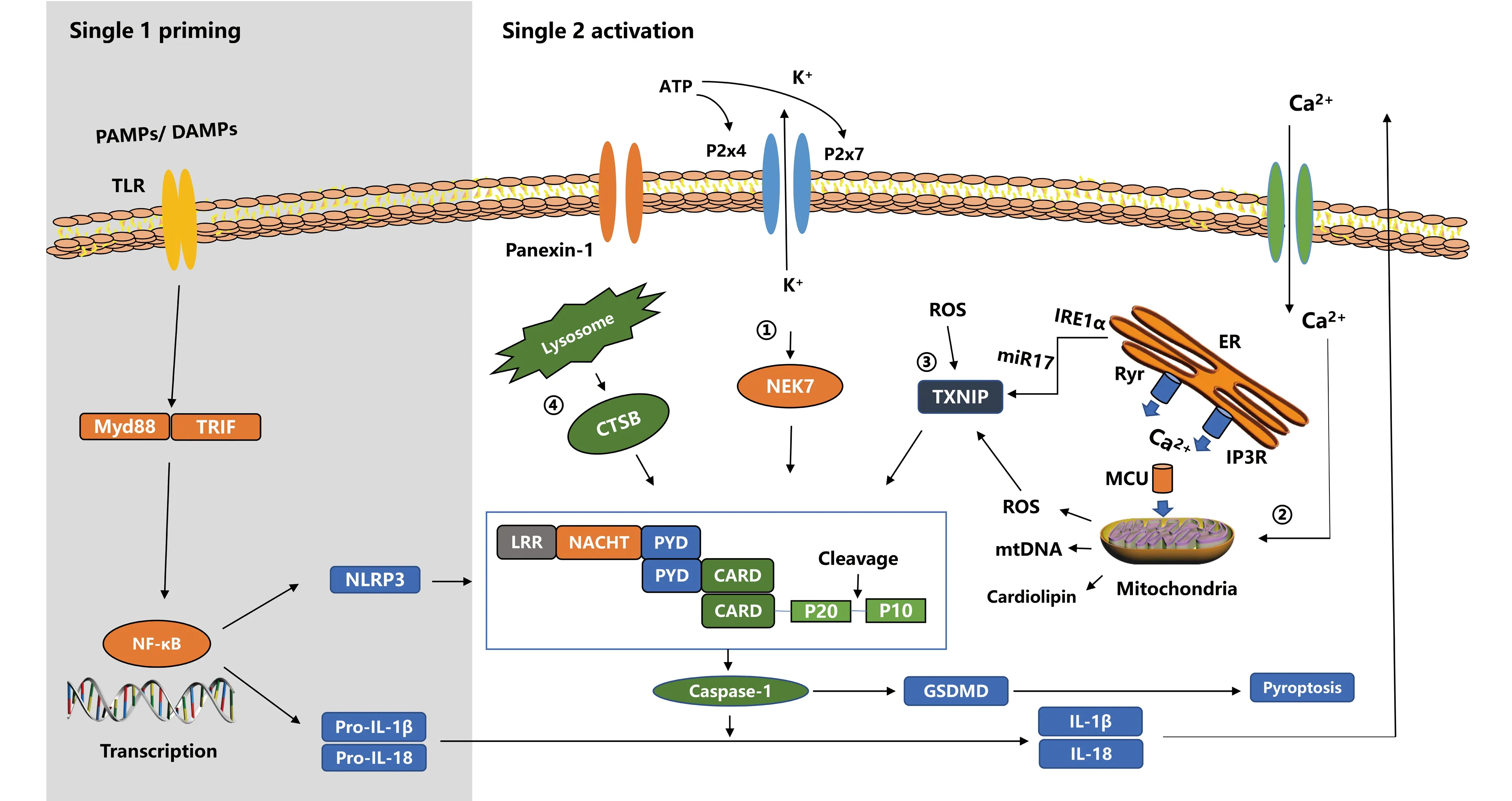
Fig.2 Activation of the NLRP3 inflammasome The priming signal(signal 1,left)is provided by PAMPs or DAMPs, leading to the activation of the transcription factor NF-κB and subsequent upregulation of NLRP3, pro-IL-1β and pro-IL-18.The activation signal(signal 2, right)is provided by multiple molecular or cellular events, including ionic flux(efflux of cytosolic K+ and influx of Ca2+), mitochondrial dysfunction, ROS generation, and lysosomal damage, which have been shown to activate the NLRP3 inflammasome
2.2.1 钾离子外流 K+外流是目前较为公认的可以引起NLRP3炎性小体激活的生物事件。损伤发生后,大量垂死的细胞释放ATP与嘌呤能受体P2X4和P2X7发生作用,激活离子门控通道,促使细胞内K+外流,随后细胞内低K+水平诱导NLRP3炎性小体激活[25],其机制目前认为可能与诱导NIMA相关激酶7(NIMA-related kinase 7,NEK7)结合NLRP3的LRR结构域,触发NLRP3炎性小体的寡聚有关[26-27]。同时,高浓度的细胞外K+会打开泛素1(pannexin-1)通道,泛素1通道是一个非选择性的膜通道,可以介导质膜通透性增加和炎性小体激活[28]。有研究发现,P2X4受体在脊髓神经元中表达,P2X4基因敲除小鼠在脊髓损伤后胱天蛋白酶1、IL-1β水平比野生型小鼠降低,炎性小体活化减少[29];脊髓小胶质细胞中的NLRP3炎性小体和胱天蛋白酶1水平会被P2X7受体的激动剂上调,被P2X7受体的拮抗剂下调,并伴随脊髓损伤后神经炎症的促进和抑制[30]。由此可知,ATP在脊髓损伤后作为DAMPs与P2X4和P2X7受体发生作用,降低细胞内K+水平,开放泛素1通道,促进NLRP3炎性小体激活。
2.2.2 线粒体钙离子超载 Ca2+超载造成的线粒体损伤也是造成NLRP3炎性小体激活的生物事件。斯里兰卡肉桂碱受体(ryanodine receptor, Ryrs)和三磷酸肌醇受体(inositol 1,4,5-triphosphate receptor, IP3R)是胞内两大Ca2+释放通道[31]。损伤发生时,胞外的Ca2+内流到胞内,内质网(endoplasmic reticulum,ER)腔中的Ca2+也通过Ryrs和IP3R途径释放到胞内,导致细胞质中游离的Ca2+水平升高,过量的Ca2+通过线粒体钙单向转运蛋白质(mitochondrial calcium uniporter, MCU)向线粒体基质中积聚,线粒体中Ca2+超载并导致线粒体损伤[32-33]。线粒体损伤后释放的线粒体活性氧(mtROS)、氧化的线粒体DNA(mtDNA)和心磷脂(cardiolipin)等都是NLRP3的激活物[34-35]。有研究报道,通过Ryrs或IP3R释放的高水平Ca2+会导致脊髓损伤后的继发性损伤[36-37],并且在脊髓损伤之后不久,Ca2+通过MCU进入了线粒体[38]。因此,Ca2+超载造成的线粒体损伤也是脊髓损伤后NLRP3炎性小体的激活因素。
2.2.3 活性氧生成与内质网应激 ROS过度生成会触发NLRP3炎性小体激活[39]。研究发现,NLRP3具有连接PYD和核苷酸结合位点域的二硫键,并且对改变的氧化还原状态敏感,ROS能通过修饰该二硫键来触发NLRP3炎性体活化[40]。此外,ROS会破坏硫氧还蛋白(thioredoxin, TRX)和硫氧还蛋白互作蛋白(thioredoxin-interacting protein, TXNIP)的结合作用,促使TXNIP与NLRP3结合激活[41]。ROS的来源较多,由NADPH酶衍生的ROS被认为是ROS的主要来源之一。脊髓损伤后,晚期氧化蛋白质产物(advanced oxidation proteins,AOPPs)可通过NADPH酶的亚型NOX4来诱导小胶质细胞产生ROS,并激活NLRP3炎性小体[19]。线粒体被认为是ROS的另一大来源,Ca2+超载损伤线粒体释放mtROS可以激活NLRP3。除此之外,内质网应激作用于线粒体也能释放ROS。ROS生成与内质网应激是互为因果、相互促进的关系。一方面,Ca2+失衡和高ROS水平都是导致内质网应激的应激因素[42];另一方面,在应激时,内质网的跨膜蛋白需肌醇酶1α(inositol-requiring enzyme1α, IRE1α)响应应激与伴侣分子蛋白Bip相解离[43],诱导miR17(一种使TXNIP不稳定的microRNA)降解来促进TXNIP表达增加,TXNIP可直接结合并激活NLRP3,易位至线粒体又会促使ROS释放[44-45]。Yanagisawa等[46]发现,在脊髓损伤的早期,除了NLRP3炎性小体的成分表达增加,TXNIP表达也升高。此外,Ren等[47]发现,凝集素-3(galectin-3,Gal-3)(一种炎症信号分子)通过ROS-TXNIP-NLRP3信号通路促进了脊髓损伤模型中的炎性小体激活。因此,在脊髓损伤后,ROS生成与内质网应激相互促进,可能通过作用于TXNIP促进了NLRP3炎性小体激活。
2.2.4 溶酶体破裂 颗粒物质,例如二氧化硅、石棉、淀粉样蛋白β、胆固醇晶体和钙晶体等,引起吞噬作用导致的溶酶体破裂也是NLRP3炎性小体激活的诱导因素之一[48-49]。溶酶体破裂释放的CTSB可能是激活NLRP3的作用靶点。研究显示,CTSB可以作为激活因子激活小胶质细胞中的NLRP3炎症小体,抑制CTSB显著降低了NLRP3的激活[50-51]。在脊髓损伤后,胞质磷脂酶A2(cytoplasmic phospholipaseA2, cPLA2)被激活,可破坏溶酶体细胞膜,释放CTSB[52-53]。因此,在脊髓损伤后,CTSB也可能是激活NLRP3的影响因素。
3 靶向抑制炎性小体的激活在脊髓损伤中的作用
近年的研究发现,通过影响炎性小体的激活因素来抑制炎性小体活化对脊髓损伤具有可观的治疗潜力。低浓度的一氧化碳(carbon monoxide, CO)早已被证实具有抗炎作用[54],并且可通过调节线粒体的生物发生和吞噬,对线粒体的应激也发挥保护作用,减少ROS的生成[55]。内源性的CO可由血红素加氧酶1(heme oxygenase-1, HO-1)分解血红素产生,Lin等[56]发现,血红素加氧酶1作用于脊髓损伤大鼠,可以抑制NLRP1炎性小体活化,减轻神经炎症,促进神经元存活。Zheng等[57]使用一氧化碳释放分子3(carbon monoxide releasing molecule-3, CORM-3)递送CO治疗脊髓损伤大鼠,发现CO可通过抑制IRE1α磷酸化降低TXNIP的表达来减少NLRP1和NLRP3炎性小体的激活,也改善了组织病理学和行为学的结果。他们通过尾静脉给药,避免了传统的肺部递送系统,显著降低了CO的毒性。基质细胞衍生因子1α(stromal cell-derived factor-1α, SDF-1α),又称趋化因子CXCL12,在局部免疫反应中发挥趋化因子的作用,其趋化作用在多发性硬化症模型中具有神经保护性[58]。在SCI模型中,Zendedel等[22]鞘内注射SDF-1α显著减少了NLRP3炎性小体激活和成熟的IL-1β、IL-18释放,其机制可能与促进脊髓损伤部位的小胶质细胞由M1表型转为M2表型,进而减少M1表型小胶质细胞释放的ROS有关。亚甲蓝(methylene blue)是一种有效的ROS抑制剂,Lin等[59]证实,在脊髓损伤模型中,亚甲蓝降低了小胶质细胞中ROS的生成和NLRP3炎性小体激活,减轻了组织损伤并促进了功能恢复。Zhou等[60]使用P2X7受体的抑制剂亮蓝G(brilliant blue G,BBG)和吴立莹等[61]使用泛素1通道特异阻滞剂丙磺舒治疗脊髓损伤大鼠,都通过影响K+外排,抑制了炎性小体的活化,减轻了脊髓损伤后的神经炎症。
通过影响炎性小体成分蛋白质和促炎因子前体的转录修饰来治疗脊髓损伤也具有一定可行性。研究证实,一些对NF-κB信号通路有抑制作用的中草药提取物,例如雷公藤红素[18]、白藜芦醇[62]和紫雏菊苷[63]等,在脊髓损伤模型中都可通过抑制NF-κB信号通路,降低小胶质细胞中NLRP3蛋白和IL-1β、IL-18前体的转录,从而减少NLRP3炎性小体活化,并最终减轻炎症反应改善功能。
对炎性小体成分蛋白质进行特异性抑制也是一个有希望的药物开发思路。MCC950是一种NLRP3的特异性抑制剂,已在多种NLRP3相关疾病中得到验证[64-65],在脊髓损伤模型中,也可以抑制NLRP3炎性小体减轻炎症反应[66]。
此外,其它一些以炎性小体作为靶标但作用机制暂时不太明确的治疗也取得了可喜的结果。褪黑素(melatonin,MT)和17β-雌二醇(17β-estradiol,E2)这2种激素都被证明有抗炎作用,都可以通过抑制NLRP3炎性小体活化降低脊髓损伤后的炎症反应,但达到相同的抗炎效应,E2所需要使用的剂量更小[67]。Mohammed等[68]与Huang等[69]分别将来自于神经干细胞的细胞外囊泡经鞘内注射和静脉给药作用于脊髓损伤大鼠,都被证实可以抑制NLRP3炎性小体的激活,减少细胞焦亡。
4 问题与展望
炎性小体在脊髓损伤后的继发性神经炎症进程中发挥着重要作用,其下游介导的细胞焦亡也被认为是脊髓损伤后细胞程序性死亡的重要途径之一。靶向抑制炎性小体的激活以减轻继发性损伤是治疗脊髓损伤的新策略。但是,炎性小体在脊髓损伤中的研究起步较晚,仍有许多未解之谜。例如,目前的研究主要涉及NLRP1和NLRP3炎性小体,其它炎性小体亚型是否也参与引发脊髓损伤后的神经炎症,尚待更多的研究来证明,在不同神经细胞中激活的炎性小体亚型仍待进一步确定。目前,一些靶向抑制炎性小体激活的治疗方法取得了可喜的结果,但距离实现临床转化仍有较长一段路程要走。目前的研究仅局限于动物实验,应用于临床仍需评估安全性,其中一些疗法显示了对炎性小体的抑制效果,但是作用机制尚不明确;一些疗法(例如激素疗法)无组织特异性,不良效应较多;一些疗法(例如CO)采用何种药物制剂和给药方式才能实现治疗作用也值得思考。因此,进一步完善炎性小体在脊髓损伤中的基础研究,并开发具有特异性的炎性小体抑制剂将是今后的研究重心。
猜你喜欢
中西医结合心脑血管病杂志(2022年19期)2022-11-19
中国临床解剖学杂志(2022年1期)2022-11-15
湖北农业科学(2022年11期)2022-07-18
检验医学与临床(2022年12期)2022-06-27
江西水产科技(2022年2期)2022-05-17
中国饲料(2022年5期)2022-04-26
实用肿瘤学杂志(2020年4期)2020-12-08
食管疾病(2020年4期)2020-12-08
食品安全导刊·中旬刊(2020年2期)2020-06-01
文苑(2018年22期)2018-11-19
