
过渡金属单原子催化剂活化H2O2/PMS/PDS降解有机污染物的研究进展
2022-08-29 04:09段毅邹烨周书葵杨柳
化工进展 2022年8期
段毅,邹烨,周书葵,杨柳
(南华大学土木工程学院,湖南衡阳 421001)
随着工业的高速发展,药品与个人护理、内分泌干扰物、染料及农药等有机污染物带来的环境风险引起国内外广泛关注,这些污染物通常具有高毒性、难降解和易累积等特点,进入水体之后会影响水生生物的正常生长和繁殖,对生态系统和人类健康构成了严重威胁。多项研究认为催化剂能够促使降解反应顺利进行,并减少二次污染物的生成,因此,为更好地应对日益恶化的水污染问题,探索和开发高性能且能广泛应用的催化剂是目前研究的重中之重。传统均相催化剂具有较高的催化活性,但存在原料和产物难以分离回收的问题,而非均相催化剂极易从反应混合溶液中分离出来,但其原子利用率却不如均相催化剂,故开发一种兼具高原子利用率和易分离回收的催化剂是研发高效催化剂的重点。亚纳米尺寸颗粒比纳米尺寸颗粒拥有更好的催化活性和选择性,而负载型催化剂金属分散的极限是将金属以单原子的形式分散到载体上,并可作为活性位点,因此,单原子催化剂(SACs)受到越来越多的关注。SACs 具有不饱和配位结构,其赋予它们独特的几何构型和电子结构。不饱和单原子作为催化反应的活性中心能大大提高原子利用率,同时单原子的同质性使其对特定反应具有很高的选择性,兼具均相催化剂和非均相催化剂的优点,在催化反应中呈现出原子利用率高、催化活性强、稳定性高及可重复利用性等特点。
单原子催化剂在多个领域展现出广阔的应用前景,包括CO 氧化、CO还原、析氢等。近年来,单原子催化剂也被应用于催化降解有机污染物,并表现出优异的催化性能。现有文献较多地综述了单原子催化剂的合成方法和表征技术,例如在高级氧化技术中SACs 的合成主要通过热解法和浸渍法,还有新兴的金属负载量较高的球磨法、熔盐法等。另外,SACs 不同于普通催化剂,对表征的要求很高,需要采用大角度环形暗场扫描透射电子显微镜(HAADF-STEM)、扫描隧道显微镜(STM)等观察和定位单个原子,直接揭示SACs的表面结构和电子结构。此外,还需X射线吸收光谱(XAS)、X射线光电子能谱(XPS)、拉曼光谱、傅里叶变换红外光谱(FTIR)和X 射线衍射(XRD)等进一步分析SACs的物理和化学性质。但对不同种类的单原子催化剂与有机污染物分子之间相互作用机理探讨较少,因此本文着重综述了Fe-SACS、Co-SACs、Mn-SACs、Cu-SACs 等单原子催化剂活化氧化剂[HO、PMS(过单硫酸盐)、PDS(过二硫酸盐)]降解有机污染物的机理,单原子金属(M)一般与N元素键合形成活性位点M—N,与氧化剂之间发生电子转移从而活化氧化剂生成·OH、SO、O等,实现有机污染物的高效降解,其概述如图1 所示。本文旨在为SACs 活化HO/PMS/PDS催化降解有机污染物的应用研究提供参考依据和指明发展方向。
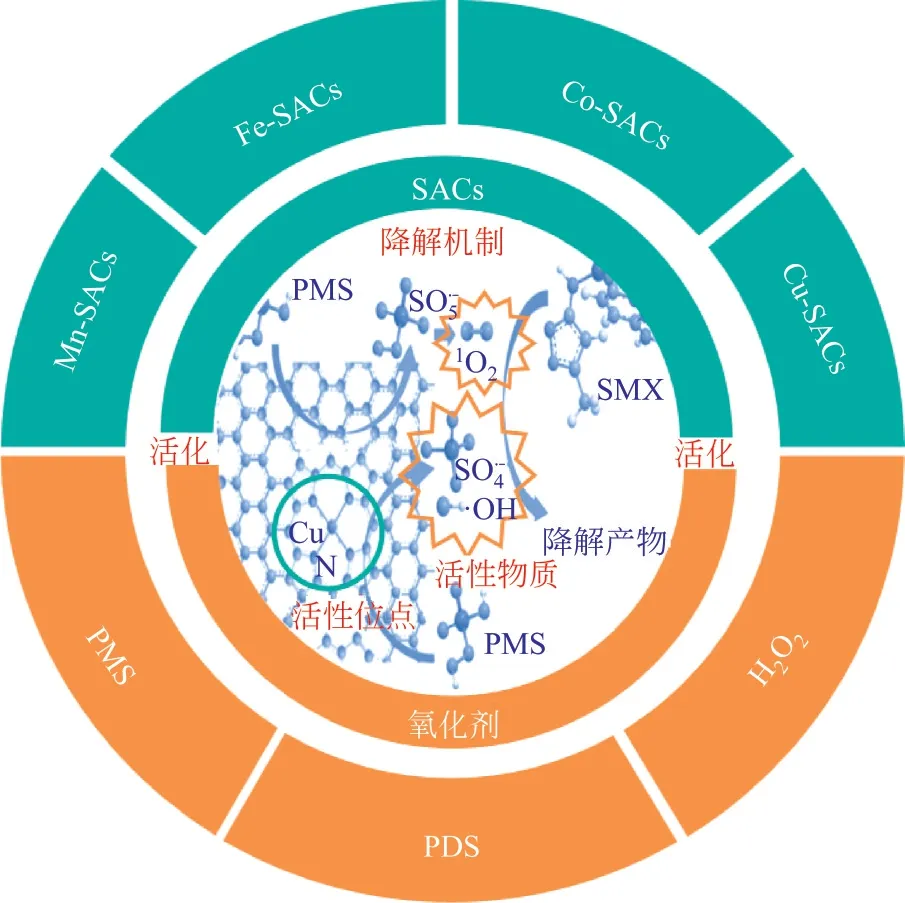
图1 不同过渡金属基SACs活化H2O2/PMS/PDS降解有机污染物的概述图
1 SACs催化降解有机污染物的机理
高级氧化体系中不同金属通常对不同氧化剂具有不一样的活化能力,特定的反应体系通常需要特定的金属催化,如Fe 基催化剂更适合HO催化,Co 基对PMS 活化效果好。为充分发展高级氧化技术(AOPs),针对目标污染物设计特定的金属基SACs显得尤为重要。
1.1 Fe-SACs
芬顿反应通常用于有机污染物的去除,但存在铁浸出、铁污泥[如Fe(OH)]形成、回收和酸碱度限制等问题,随后负载型Fe 催化剂广泛应用,非均相类催化剂被认为可解决这些问题,但活性却没有均相催化剂高。大量Fe 基材料被用于活化HO催化降解有机物污染物,降解效率高且无二次污染,但普通Fe 基催化剂由于金属位点的自然聚集趋势,导致活性位点减少。Fe-SACs的金属活性位点可达到原子级,其高度分散性提高了原子利用率,催化活性高,又能解决铁浸出、铁污泥、酸碱度限制等问题,因而被广泛用于活化HO催化降解有机污染物。Fe 通常与碳氮材料结合形成Fe-SACs,单原子Fe 通过配位作用与N 形成均匀分散的Fe-N-C活性位点,不仅原子利用率高,且可以充分活化氧化剂,从而提高催化反应速率。韩旭等将聚酞菁铁(FePc)负载在片状石墨氮化碳(ECN)载体上制备出ECN@FePc-600-2,用其活化HO降解罗丹明(RhB)。该催化剂的单原子Fe和N 配体之间相互作用强且均匀分散,促使Fe 负载量高,较多的活性位点在ECN表面上原位生长,再通过XPS光谱可知,ECN@FePc-600-2催化剂具有72%的Fe(Ⅱ)和28%的Fe(Ⅲ),它们与HO反应生成的·OH将有机物矿化。Yin等采用球磨法将Fe(NO)负载在介孔二氧化硅(SBA-15)上制备出SAFe-SBA,用其活化HO去除血红蛋白A(HBA)和苯酚。Fe 原子将SBA-15 载体上Si—OH 的四个氢原子取代,形成均匀分散的Si—O—Fe 活性位点,促使Fe 负载量高,并提高了Fe 原子的利用率,该活性位点与HO反应生成大量的·OH[式(1)、式(2)],降解有机污染物[式(3)]。

Fe-SACs还可以活化过硫酸盐催化降解有机污染物,利用Fe与Fe循环产生SO、O或高价铁氧化有机污染物。碳氮基Fe-SACs 在活化PMS 的过程中,高电子密度的氧原子可以作为电子供体传递电子,将PMS 还原为·OH 和SO;邻近低电子密度的C 原子作为电子受体,PMS 作为电子供体,电子从PMS转移到C原子产生SO,随后与水分子反应生成O。即在缺电子和富电子位点上分别与PMS 发生氧化和还原反应,同时产生O和自由基(·OH 和SO),增强有机污染物的降解。Yao等将Fe 嵌入共价有机骨架(COF) 中制得Fe@COF,用其活化PMS 降解金橙Ⅱ(AO7)。单原子Fe 通过配位作用与N 形成均匀分散的Fe—N活性位点,并以多层导电的多孔碳材料为载体,形成大量的Fe—N—C 活性位点,PMS 分子首先与Fe—N—C 位点成键,PMS 和C 原子分别作为电子供体和电子受体,它们之间的电子转移促使PMS有效活化并产生大量O,高效降解AO7,见图2。PDS 的活化机理与PMS 类似,Jiang 等采用球磨法制备了单原子Fe-N-C 复合物,用其活化PDS 去除2,4-二氯苯酚(2,4-DCP)。PDS首先吸附至活性位点(Fe—N键),从Fe(Ⅲ)中获得两个电子而发生还原分解,并将Fe(Ⅲ)氧化成Fe(Ⅴ)或Fe(Ⅴ)=O,从而高效降解有机污染物。Huang 等采用水热解法制备出FeMoS,利用其活化PDS 去除普罗奈尔(PPA)。该催化剂在pH 为3~8 时具有较高的催化效率,这是由于催化剂表面MoS纳米片边缘存在大量不饱和S 捕获溶液中的H,使催化反应能在较大的酸碱度范围内进行。以Fe 原子和Mo 原子作为活性中心,通过Fe与Fe循环,以及Mo与Mo、Mo循环,催化PDS 生成SO、SO、SO、·OH,Fe 的高利用率和Mo 活性位点的协同作用共同提高了PDS 的活化性能,从而高效降解PPA。
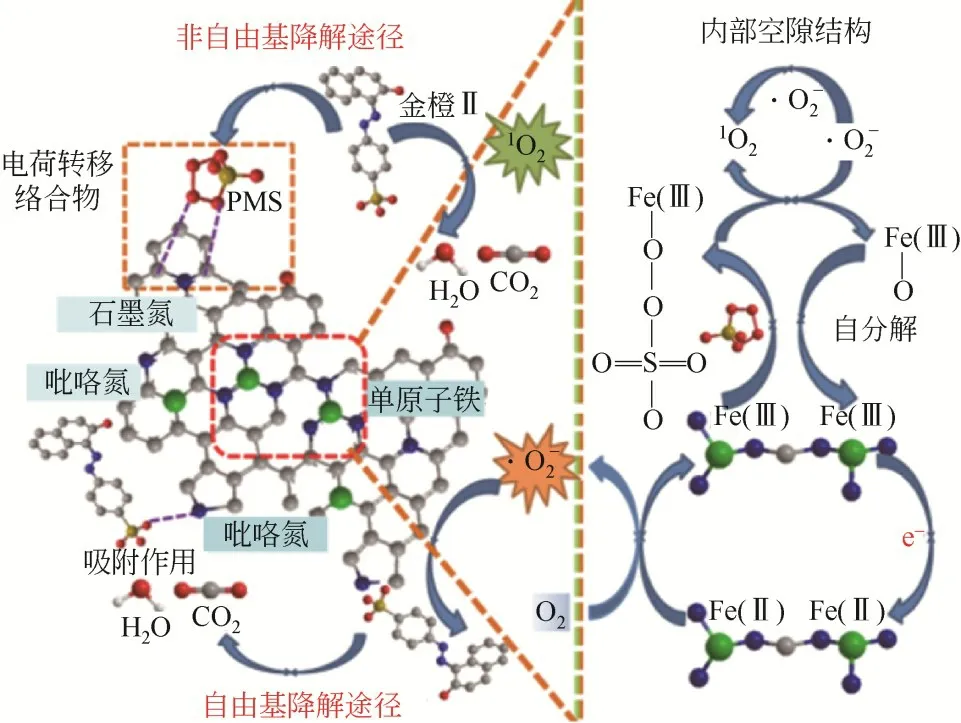
图2 Fe@COF活化PMS降解AO7[19]
1.2 Co-SACs
Co被认为是活化PMS 的最佳金属活化剂,Co-SACs 已被大量研究,通常将Co 负载于CN 材料,单原子Co与N形成均匀分散的Co—N配位体,作为催化反应的活性中心,用其活化PMS 高效去除含不饱和键和芳香环的有机污染物。Co-SACs催化剂通常以表面均匀分散的单原子Co 作为活性位点,与Fe-SACs 类似,通过与PMS 发生电子转移产生O,或通过Co与Co循环产生SO,降解有机污染物。Chen等采用热解法制备出单原子催化剂Co-N-C,用其活化PMS 去除双酚A(BPA),吸附和催化性能优异。BPA 首先被吸附预浓缩在Co-N-C 表面,随后PMS 与均匀分散的活化中心Co-N结合,促使PMS 之间发生电子转移产生O[式(4)],从而催化吸附的BPA 氧化降解为CO和HO(见图3),释放的活性位点再次吸附溶液中的BPA,进入新的吸附和催化降解循环。徐劼等采用模板蚀刻法合成单原子催化剂Co-C-N,用其活化PMS 去除AO7。均匀分散在石墨烯载体表面的Co 原子为活化中心,与其键合的C 原子和PMS 分别作为电子受体和供体,电子从PMS转移到C原子以产生SO,随后SO自由基与水分子反应生成O,它们共同降解AO7。Chu 等将Co 嵌入一种四吡啶(TPML) 制备出单原子催化剂Co-TPML,用其活化PMS 降解BPA。一方面该催化剂表面均匀分散的Co 原子与N 形成Co—N配位的活性位点,与PMS发生电子转移从而产生SO/·OH;另一方面,TPML 载体上的芳香环离域π 电子会促进PMS 的O—O 键分裂,使表面稳定产生·OH,上述两方面的协同作用促使BPA高效降解。

图3 Co-N-C活化PMS降解BPA[24]
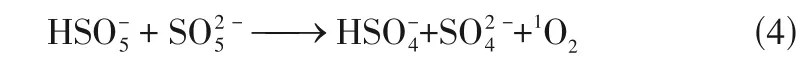
1.3 Mn-SACs
Mn-SACs 内部稳定分散的Mn单原子与在催化反应过程中浸出的Mn都能活化PMS 实现有机污染物的高效去除,但金属Mn 的稳定性强,Mn-SACs 与氧化剂之间的反应活性要弱于Fe-SACs 或Co-SACs。Mn-SACs 通常是将Mn 与碳氮材料结合,形成均匀分散的Mn—N活性位点,可与氧化剂充分接触,提高催化活性,广泛应用于去除持久性有机污染物。Yang 等将Mn 锚定在氮掺杂的多孔炭上制备单原子催化剂Mn-ISAs@CN,用其活化PMS 去除BPA。PMS 首先吸附至均匀分散的Mn—N活性位点上,两者之间相互作用产生·OH和SO,其次,BPA 通过电子供体-受体机制被吸附至吡咯氮上(毗邻Mn—N),因此,原位生成的活性自由基可以迅速与邻位吸附的污染物发生反应,从而显著提高催化效率。此外,催化反应过程中还发现浸出少量的Mn,也可以作为催化剂降解BPA(7%)。Guo等采用热解法将Mn负载在氯化萘(CN)上制得单原子催化剂Mn—CN,用其活化O和HO去除草酸(OA),即便是在酸性(pH=3)条件下也能高效降解OA。均匀分散的催化活性位点Mn—N首先与HO作用形成HOO—Mn—N,随后与O反应生成HO·和O,之后O在酸性溶液中立刻与H结合形成HO·,并迅速转化为·OH,从而实现OA 的高效去除,见图4。柯倩采用研磨-高温煅烧法将Mn 负载于氮掺杂石墨烯(NG)制得单原子催化剂SA-Mn/NG,用其活化PDS 高效去除抗生素磺胺甲噁唑(SMX)。实验结果表明,·OH 和SO是降解SMX 的主要活性自由基,基于Mn与Mn循环及O参与反应,提出了SAMn/NG+PDS 体系中SMX 的降解机理,如式(5)~式(11)。
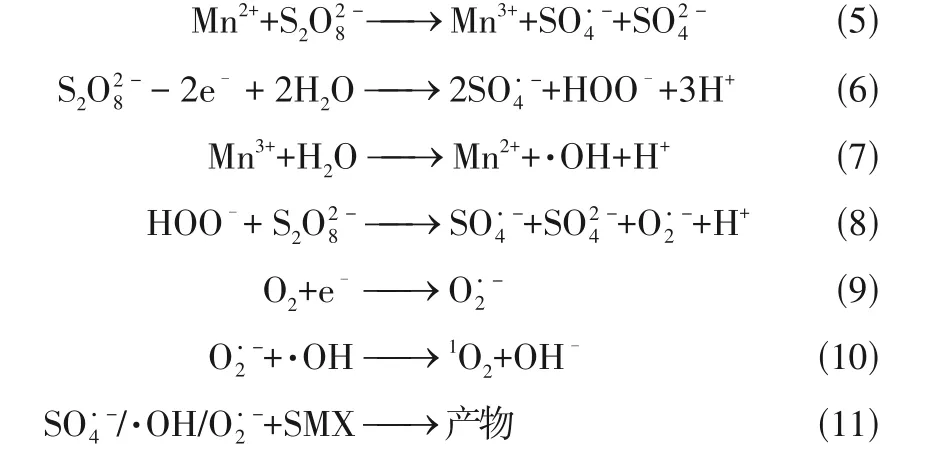
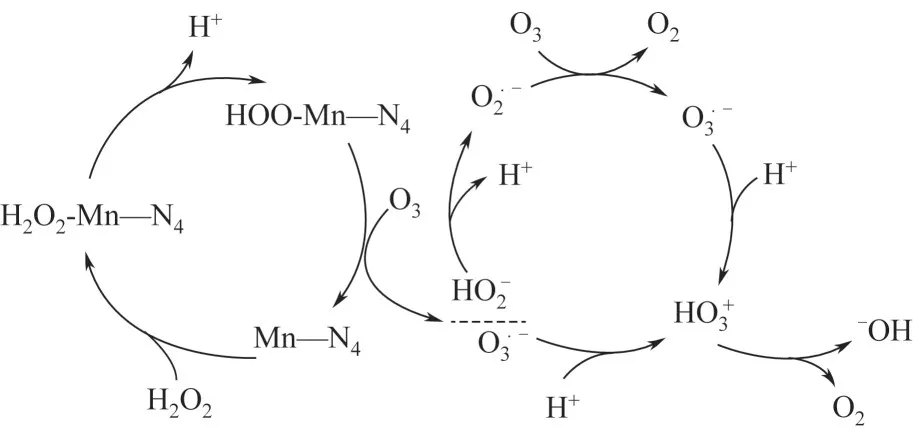
图4 Mn-CN活化H2O2降解OA[29]
1.4 Cu-SACs
Cu作为SACs 金属中心催化有机污染物越来越受到关注。Cu-SACs也是将Cu负载在碳氮材料上,形成作为活性中心的Cu—N配位体,有效活化氧化剂,产生大量自由基,高效降解有机污染物。Xu 等采用热解法制备出单原子催化剂Cu-CN,用其活化HO降解RhB。HO首先被吸附至Cu—N位点并成键,HO和缺陷C原子分别作为电子供体和电子受体,它们之间的电子转移促使HO有效活化并产生大量·OH,且以多层导电的多孔碳材料为载体,促使HO与内部活性位点充分接触,实现RhB的高效降解。Chen等采用球磨法将Cu负载在氧化石墨烯(GO)上制备出SA-Cu/rGO,用其活化PMS 去除SMX。均匀分散在GO 上的Cu 单原子与PMS 充分接触,它们之间发生电子转移产生·OH 和SO[式(12)]。此外,电子从PMS上转移至缺陷C 原子上也会形成SO[式(13)],与水反应生成O[式(14)],多种自由基的协同作用实现了SMX的高效降解,见图5和式(15)。

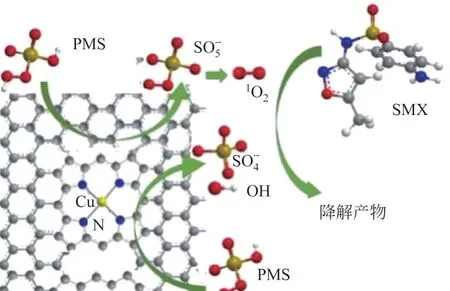
图5 SA-Cu/rGO活化PMS降解SMX[32]
1.5 双金属SACs
目前双金属单原子催化剂较多地应用在二氧化碳还原、氧还原/氧析出等领域,催化活性较好。双金属SACs 在去除有机污染物的领域也有应用,主要通过双金属单原子的电子结构优化或通过某一金属促进另一金属再生的方式,与氧化剂发生电子转移产生SO、·OH 和O,降解有机污染物。邓方鑫采用热解法以铁掺杂的沸石咪唑骨架材料(Fe-ZIF-8)为原料制备出Co/Fe-N-C,用其活化PDS 去除苯酚,在较宽的pH(pH=3~9)范围内均能对苯酚有效矿化。一方面,该催化剂表面有大量均匀分散的Co、Fe原子活性位点,原子利用率高,与PDS 之间发生电子反应,产生大量的SO;另一方面,电子从PDS 转移至缺陷C 原子上形成SO,与水反应生成O,上述自由基的协同作用强化苯酚的去除。Chen 等以铁、铋-有机框架(Fe、Bi-MOFs)为原料,采用热解法制得FeBi-NC双金属单原子催化剂,用其活化PMS降解RhB,10mg/L RhB 在较宽的pH(pH=4~10)范围内均能被完全降解。这是由于活性位点Fe-N在双金属单原子催化剂FeBi-NC中比在单金属原子催化剂中具有更强的吸附能和更长的(HO—OSO),有利于吸附PMS,形成SO和·OH。同时PMS 与氮杂化的碳环之间发生电子转移形成SO,与水反应生成O,与SO和·OH 共同氧化降解RhB。Wu等采用热解法以铁/铜-沸石咪唑骨架材料(Fe/Cu-ZIF-8)为原料制得Fe/Cu-N-C 双金属单原子催化剂,用其活化PDS 降解氯霉素(CAP),相比于单金属原子催化剂Fe-N-C,CAP 的催化速率从0.073min提升至0.093min。这是由于Cu 元素的引入优化了Fe 3d 轨道的成键轨道的分布,促进了PDS的吸附和裂解,生成SO、·OH、O,从而高效降解CAP。
上述双金属单原子催化剂的活性得到提升,主要是因为引入不同单原子可以优化催化剂的电子结构,从而提高催化性能。此外,双金属单原子催化剂还可以通过某一金属促进另一金属再生的方式,提高单原子催化剂的活性。梁言等采用热解法制得单原子催化剂Fe-Ce/g-CN,用其在可见光下活化HO去除亚甲基蓝(MB)。Ce/Ce和Fe/Fe的标准氧化还原电势分别为1.44V 和0.77V,热力学上有利于电子从Fe向Ce转移[式(16)],故反应体系可以稳定地提供Ce与HO之间发生电子转移,产生更多的·OH,从而高效地去除MB,见图6。
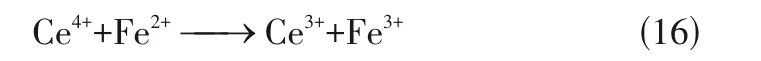

图6 可见光下Fe-Ce/g-C3N4活化H2O2降解MB[36]
1.6 其他SACs
陈枫将Cr负载在g-CN上制备出单原子催化剂SA-Cr/g-CN,在可见光的照射下用其活化HO降解BPA,在较宽的pH(pH=3~11)适用范围具有较高的降解和矿化效率。这是因为SA-Cr/g-CN有大量均匀分散的活性位点Cr—N,可有效活化HO生成·OH。另外,暴露的单原子Cr(Ⅱ)可通过光生电子有效再生,原子利用率增加。Huang 等采用浸渍法以SiC 为载体合成了单原子催化剂Pt/SiC,对全氟辛烷磺酸(PFOA)催化活性高。这是因为单原子Pt可以降低H原子的吸附能、影响Pt/SiC 能带结构,从而在紫外线的作用下加快形成Pt—H键,单原子Pt还可以加快H原子的溢出,与SiC表面形成Si—H键,最后与C—F键进一步转化为Si—F/C—H,从而高效催化PFOA。Feng 等采用热解法制得单原子催化剂Pt/AlO,用其活化PMS 降解1,4-二氧己环(1,4-D),去除率超过95%。该催化剂表面均匀分散的Pt与PMS通过均裂反应产生大量的表面络合SO,转化率几乎达到100%,从而高效去除1,4-D。Wang 等将Ag 负载在g-CN上制备出单原子催化剂Ag/mpg-CN,用其在可见光下活化PMS高效降解BPA,见图7。该催化剂上均匀分散的Ag 单原子通过表面等离子共振(SPR)使材料对可见光的吸收能力提高,并且SPR效应还可以增强局域电磁场,从而加快e和的生成速率[式(17)],PMS 与e结合产生SO[式(18)],此外,e与O反应形成O[式(19)],与h共同氧化BPA[式(20)]。

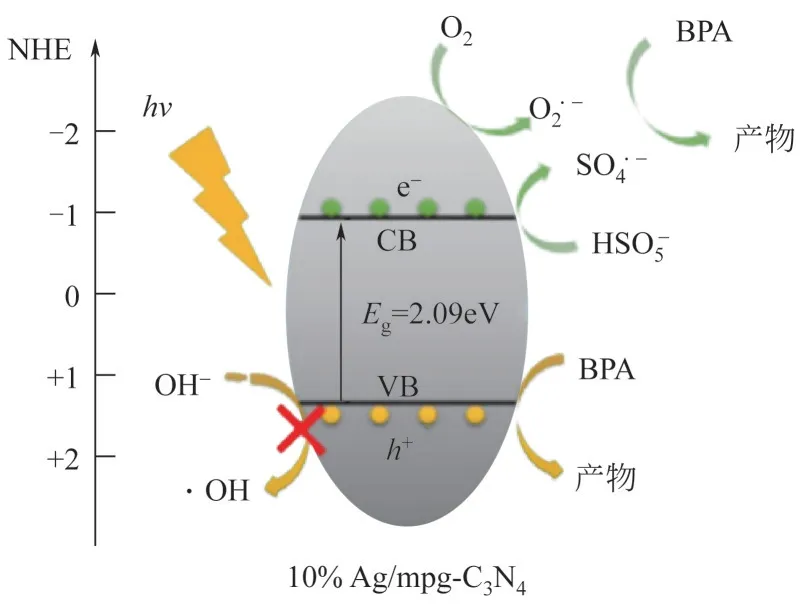
图7 可见光下Ag/mpg-C3N4活化PMS降解BPA[40]
综上所述,单原子催化剂基本上都是以碳氮材料为载体,过渡金属(M=Fe、Co、Mn、Cu等)与N键合形成活性位点M—N,活化HO/PMS/PDS高效去除有机物污染物,其机理类似,都是依靠M与M之间的转化[式(21)~式(23)]不断产生活性自由基,或单原子催化剂的活性位点与HO/PMS/PDS 通过供体受体机制实现电子转移产生O,实现有机污染物的高效降解。此外,Fe-SACs还可以通过生成Fe(Ⅴ) ==O氧化有机污染物。然而,不同过渡金属单原子催化剂活化氧化剂的种类不同:Fe-SACs 可以活化HO/PMS/PDS 实现有机污染物的高效降解;Co-SACs 一般只用于活化PMS/PDS,且相对于PMS,活化PDS 降解有机污染物的效率低;Mn-SACs 和Cu-SACs 一般用于活化PMS/HO降解有机污染物,很少用于活化PDS。双金属单原子催化剂可以加速金属的价态转换或优化电子结构,此外单原子催化剂还可以引入光催化,都可加速有机污染物的降解,但这方面的研究机理较少,值得进一步深入探索。
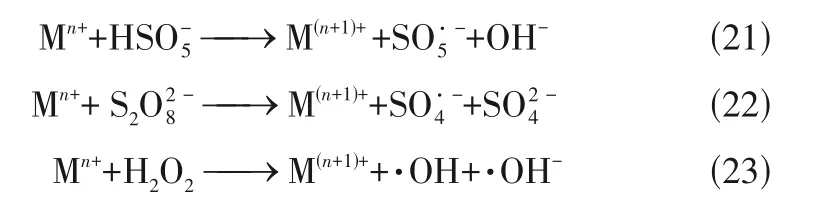
2 SACs 催化降解有机污染物的影响因素
SACs 催化降解有机污染主要的影响因素有金属负载量、催化剂投加量、氧化剂投加量、pH、有机物初始浓度、温度和无机阴离子。
2.1 金属负载量
Qi 等研究了不同Co 负载量下的单原子催化剂SA Co-N/C 活化PMS 对萘普生(NPX)的降解效率,实验结果表明,不同金属负载量的催化剂对NPX 的降解效率不同,当Co 负载量由0.26%(质量分数)增加至2.35%(质量分数)时,NPX的降解效率随之增加,当继续增加至3.9%(质量分数)时,降解效率反而降低,这可能是由于过高的负载量造成单原子团聚;当Co 负载量为2.35%(质量分数)时,50min 内NPX 的降解率最佳达到100%。Zhang 等采用高温煅烧法制备出不同Fe 负载量的SA Fe@NSC,利用其活化HO降解甲砜霉素(TAP),实验结果表明,Fe 的负载量质量分数为0.45%、0.77%、1.62%、2.04% 时,40min 时TAP 的降解效率分别为70%、90%、99%、93%。因此,单原子催化剂的金属负载量还有很高的提升空间。
2.2 投加量
Yang 等采用热处理法制得单原子催化剂Fe-N/C,在不同催化剂投加量下活化PMS 降解BPA 的实验中发现,当催化剂投加量由0.1g/L 增加至0.15g/L 时,20min 时BPA 的降解效率由91%增加至99.3%,但当投加量进一步增加到0.25g/L时,10min 时降解率就达到了100%,这是由于更多的催化剂提供了更多的活性位点。Chen 等将Cu 负载在氧化石墨烯(GO)上制得SA-Cu/rGO,研究其在不同的氧化剂浓度下对SMX 降解效率的影响,当PMS 浓度从0.2g/L 增加至0.4g/L 时,60min 后SMX 的降解效率由63.0%增加至99.6%,但浓度继续增至0.5g/L 时,SMX 降解率却略有下降。这可能是加入过量的PMS 会产生过量的SO,导致SO发生自淬灭反应。因此,催化剂和氧化剂的投加量都有一个最佳值,过多或过少都不利于催化反应。
2.3 pH
徐劼等在Co-C-N 活化PMS 降解AO7 的实验中,酸性条件下反应速率受到轻微抑制,完全去除AO7 的反应时长由10min 延长到30min,而在pH=9.0时,PMS被碱活化生成O去除AO7,反应速率迅速得到提升。Zhang 等采用热解法制得Fe/CN,用其活化PMS降解4-氯苯酚(4-CP),在探究不同pH 对4-CP 降解效率的影响中,发现pH 为8 时降解效率最好,60min 时4-CP 几乎完全降解,但随着溶液pH 升高至11,催化活性逐渐降低,60min时4-CP 的降解效率降低至94%。柯倩在不同pH下SA-Fe/g-CN活化HO降解BPA 的试验中发现,当pH≤4 时,SA-Fe/g-CN均表现出较强的催化活性,BPA 在40min 内几乎降解完全;当pH=2.5 时,其催化活性最高,30min内BPA几乎完全降解,但随着溶液pH 的升高,催化活性逐渐降低,表明SA-Fe/g-CN活化HO降解BPA过程适合在酸性条件下反应。上述研究表明,SACs 活化过硫酸盐催化降解有机污染物需要溶液初始pH 为弱碱条件,活化双氧水需要酸性条件,这与大多数研究的结论一致,因此,拓宽SACs的pH应用范围还值得进一步探索。
2.4 有机物的初始浓度
Pan等采用高温煅烧法制得SA Cu@NBC,用其活化PMS 降解BPA,发现BPA 的初始浓度为20mg/L 时,降解效率最高,但随着BPA 的初始浓度增加,降解效率有所降低,这可能是由于活性位点不够导致的。Zhao 等用高温煅烧法制得BCN/CoN,用其活化PMS降解四环素(TC),当TC的初始浓度由25mg/L 增加至50mg/L 时,降解效率由98%增加至100%,但当继续增加至70mg/L 时,降解效率反而下降至89%。这可能是由于TC 初始浓度过高时活性位点过少导致的。
2.5 温度
Yin 等探索在不同的温度下SAFe-SBA 活化HO去除HBA,发现高温有利于HBA 的去除,在25℃下降解100% HBA 需要90min,在35℃下需要60min,在45℃下只需要30min,这可能是活性氧浓度随着温度升高而增加,导致HBA降解速度加快。韩旭等研究在不同的反应温度下,ECN@FePc-600-2单原子催化剂对RhB降解效率的影响,发现相对于30℃和40℃,在10℃和20℃时RhB的降解速率较低,表明温度对降解效率有一定的影响。
2.6 无机阴离子
Peng等采用热解法制得SA Co-N-C,用其活化PMS降解氯霉素(CQP),发现Cl和NO对CQP的降解效率几乎无影响,但HPO在浓度较低时会促进PMS 的分解,促使更多的活性基团产生,这是由于结构不对称的PMS 易受亲核物质HPO-攻击。但当HPO的浓度增加至10mmol/L时,CQP的降解效率略有抑制,这可能是HPO-吸附在SA Co-N-C 表面导致。Yao 等探究Fe@COF 活化PMS 降解AO7反应过程中无机阴离子对催化反应的影响,发现Cl和HPO对降解效率的影响极小,而HCO的浓度从5mmol/L增加至40mmol/L时,降解效率从100%下降到65.7%,显著抑制了AO7 的降解,这可能是由于反应过程中HCO与主要的活性基团O产生了反应,消耗了一部分O,从而抑制了降解效率。无机阴离子对单原子催化剂降解有机污染物的影响随催化剂和氧化剂种类变化而发生变化,宜具体情况具体分析。
综上所述,金属负载量、催化剂或氧化剂投加量、pH 对催化反应的影响较大,而温度、无机阴离子对催化反应的影响较小,因此,为使SACs 高效催化降解有机污染物,首先需着重考虑金属负载量、投加量和pH等影响因素。
3 SACs催化降解有机物的应用
SACs 在环境应用中的性能很大程度上受其性质(如金属原子的配位状况,与反应物和中间体的吸附及相互作用,金属、载体和反应物之间的电荷相互作用)影响。SACs有较强的金属-配体相互作用力和电荷转移距离短等特点,使其具有较高的原子利用率,电荷分离也得到改善,结合载体材料的协同作用,都证明了SACs 的催化性能优于其纳米颗粒。如将金属单原子负载到半导体(如g-CN)上,可增强原子利用率、电荷转移速率、自由基生成速率。目前,许多研究者将这些发现应用于催化降解有机污染物,以修复水生态环境。本文将近年来报道的单原子催化剂应用于降解有机污染物的研究总结于表1,并以活性中心单原子金属进行分类,为单原子催化剂在水处理领域的应用提供参考。
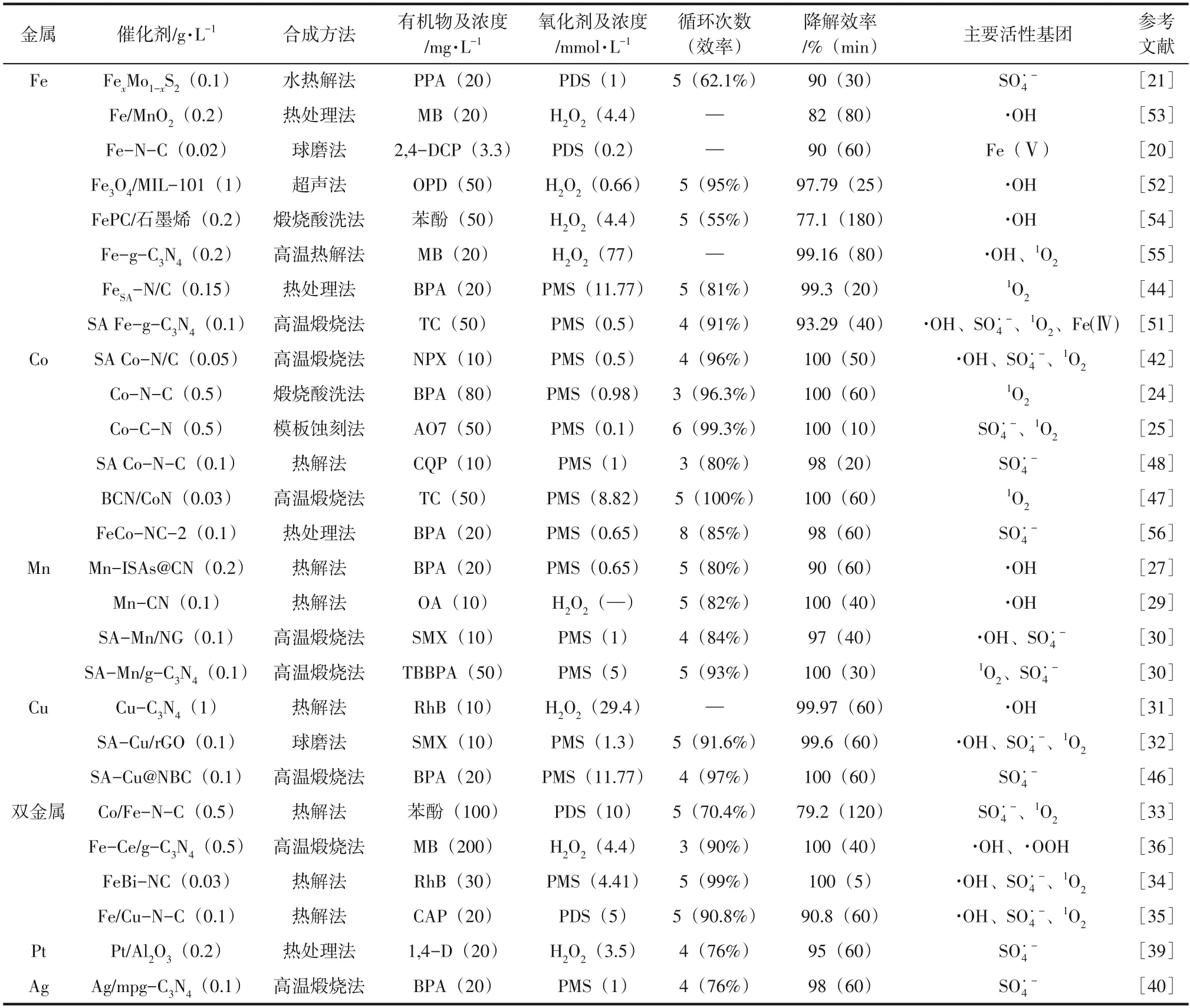
表1 单原子催化剂用于高级氧化中降解有机污染物的应用
3.1 Fe-SACs
Peng 等采用高温煅烧法制得SA Fe-g-CN,用其活化PMS降解TC,40min时降解率为93.29%,经过4 次循环实验后,降解率仍有81%。且将4 次循环试验后的催化剂置于N环境下,在350℃进行热处理3h后,催化剂的活性可以恢复大部分。Yin等采用球磨法制得SAFe-SBA,用其活化HO去除HBA,90min时降解率为100%,经过3次循环实验,180min 时降解率依次为90.6%、49.0%,将其进行热处理回收后,180min 时降解率又为99.5%。Fe-SACs 除可有效降解TC 和HBA 以外,还可高效降解普罗奈尔(PPA)、2,4-DCP、邻苯二胺(OPD)、MB、苯酚和TAP等。
3.2 Co-SACs
Qi 等采用高温煅烧法制备了SA Co-N/C,用其活化PMS 降解NPX、环丙沙星(CIP)、甲硝唑(MNZ),50min 时三种有机物的降解率均为100%,并且经过4 次循环后,NPX 的降解率仍有97%。Zhao等采用高温煅烧法制得BCN/CoN,用其活化PMS 降解TC,60min 时降解率为100%,经过5 次循环实验后,降解率仍然没有降低。Co-SACs除可有效降解NPX、CIP、MNZ 和TC 以外,还可高效降解BPA、AO7等。
3.3 Mn-SACs
Yang等采用热解法制备出Mn-ISAs@CN,用其活化PMS 去除BPA。60min 时降解率为90%,每次循环前将催化剂在氩气氛中于650℃退火处理,经过5 次循环后,降解率仍有80%。Guo 等采用热解法制得Mn-CN,用其活化O和HO去除OA,OA完全降解需要40min,经过5次循环后,降解率仍有82%。Mn-SACs 除可有效降解BPA 和OA 以外, 还可高效降解SMX 和四溴双酚A(TBBPA)等。
3.4 Cu-SACs
Pan等采用高温煅烧法制得SA Cu@NBC,研究其活化PMS降解BPA。60min时BPA的降解率达到100%,经过4 次循环后,降解率仍有97%。Chen等采用球磨法制备出SA-Cu/rGO,用其活化PMS去除SMX。与纯rGO相比,SA-Cu/rGO表现出较强的催化活性,60min 时,SMX 的降解率从73.5%增加至99.6%,5 次循环实验后降解率仍有91.6%,表明SA-Cu/rGO 的重复利用性好。Cu-SACs 除可有效降解BPA 和SMX 以外,还可高效降解RhB等。
3.5 双金属SACs
邓方鑫采用热解法制备出Co/Fe-N-C,用其活化PDS 去除苯酚,在较宽的pH(pH=3~9)范围内均能对苯酚有效矿化。相比于纳米材料Co NPs/Fe-N-C(56.7%),单原子催化剂Co/Fe-N-C 活化PMS 降解苯酚,120min 内苯酚降解率达到79.2%,并且经过5 次循环实验后,降解率仍有70.4%。梁言等采用热解法制得Fe-Ce/g-CN,用其在可见光下活化HO去除MB。40min 内能完全降解MB,经过3次循环后,降解率仍有90%。Chen等采用热解法制得FeBi-NC,用其活化PMS 降解RhB。5min 时RhB 的降解率达到100%,并且经过5 次循环后,RhB 降解率仍有100%。而单一金属负载SACs(Fe-NC 和Bi-NC)在5min 时RhB 的降解率稍低,分别为97%、93%。Wu 等采用热解法制得Fe/Cu-N-C,用其活化PDS 降解CAP,60min 内CAP 的降解率达到90.8%,经过3 次循环后,降解率仍然不变,而单一的Fe-N-C 在60min 时CAP 的降解率仅为64.1%。
3.6 其他SACs
Feng 等采用热解法制得单原子催化剂Pt/AlO活化PMS降解1,4-D,反应进行60min后能达到95%降解率,经过4 次循环后,降解率为76%。Wang 等采用热解法制备得到Ag/g-CN,用其活化PMS降解BPA,60min后降解率达到98%。
综上,不同过渡金属(Fe、Co、Mn、Cu 等)的SACs 活化氧化剂降解有机污染物均具有较高活性,有机污染物的降解效率高,经几次循环实验后,有机污染物降解效率较第一次有所降低,但大部分SACs 在进行热处理后,其催化活性能较大程度地恢复,应用于水环境中去除有机污染物具有广阔的空间。但SACs 对新兴的环境污染物(药物与个人护理品、内分泌干扰物、全氟化合物等)的催化降解还不够全面,针对目标污染物设计特定SACs催化剂也值得进一步探索。
4 结语与展望
SACs 作为一种新型催化剂处理水中难降解有机污染物,表现出较大的竞争潜力,但在AOPs 中使用的SACs,其制备方法都是通过减少金属前体的加入量来实现的,导致金属负载过低,并且现有SACs 中的大部分载体是与氮、碳元素配位的碳材料,催化剂的表面性质随反应时间延长而发生变化。例如,载体中吡咯氮、吡啶氮和石墨氮的分布可以改变SACs 的金属单原子与N 元素的配位数,从而影响其催化活性,虽然热处理能较大程度恢复催化活性,但循环使用次数多后导致催化活性降低,从而使SACs 在AOPs 应用中寿命不长,鉴于此,SACs用于AOPs中去除有机污染物在国内的研究还处于初期阶段。
目前,SACs 在去除有机污染物的应用中主要存在以下问题:①开发现有合成方法制备出的SACs金属负载量较低;②大多数负载的金属为Fe、Co、Cu、Mn,忽略了其他过渡金属(如Zn、Pt、Pd等),在过渡金属负载种类上还有很大的研究空间;③酸性水溶液可能会导致SACs 的金属浸出,浸出的金属会造成环境污染,且SACs 的催化性能也会下降;④大部分催化降解研究对象是较易脱色的染料和易降解的有机污染物,对其他难降解新型有机污染物的研究较少;⑤SACs 在有机污染物处理中的催化性能及其对氧化剂的作用机制、活化机理等都需要进一步明确,其应用于水处理领域的作用机理和环境影响因素还需深入地探索和完善。
因此,SACs 活化氧化剂降解有机污染物的研究还有以下几点值得探索:①开发单原子催化剂合成的新方法,以实现高金属负载量的SACs,如球磨法、熔盐法等;②丰富负载的过渡金属,继续开发稳定性高、pH适用范围更广的新型SACs,如双金属单原子催化剂等;③单原子催化剂-HO/PMS/PDS体系中引入光催化值得进一步深入探索;④对新兴的环境污染物(药物与个人护理品、内分泌干扰物、全氟化合物等)给予更多关注,同时也应考虑在实际废水处理中多种环境因素对催化降解的影响;⑤应多着眼于SACs在结构-性能关系和催化机理的研究,针对目标污染物设计特定的催化剂。随着SACs 种类的不断发展及催化机理研究的不断深入和完善,未来将实现在原子尺度上经济绿色地降解有机污染物,为SACs 在水处理领域提供广阔的应用空间。
猜你喜欢
中国农业科学(2022年16期)2022-09-19
中国农业科学(2022年15期)2022-08-09
当代陕西(2022年5期)2022-04-19
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
科学与财富(2021年33期)2021-05-10
电脑报(2020年40期)2020-11-06
电脑知识与技术(2018年19期)2018-11-01
丝绸(2018年12期)2018-09-10
