
蓖麻油基热固性不饱和聚酯材料的制备与性能
2022-08-26 08:26雷秦阳翟梦姣白莹雪张跃宏
高分子材料科学与工程 2022年6期
雷秦阳,翟梦姣,白莹雪,吕 斌,张跃宏
(1. 陕西科技大学轻工科学与工程学院,陕西西安 710021;2. 西安市绿色化学品与功能材料重点实验室,陕西西安 710021;3. 中国石油长庆油田分公司西安长庆化工集团有限公司,陕西西安 710018)
热固性不饱和聚酯是一种常见的高分子材料,主要由不饱和聚酯基体树脂(通常是由饱和二元酸和不饱和二元醇或者不饱和二元酸和饱和二元醇通过缩聚反应制得)与活性稀释剂交联固化形成的一种具有三维交联网络结构的材料。交联固化后,这种材料表现出优异的尺寸稳定性、热力学性能、耐化学腐蚀等,广泛应用于能源、化工、交通及建筑等领域[1,2]。传统的不饱和聚酯材料通常是由65%左右的不饱和聚酯基体树脂和35%的苯乙烯活性稀释剂两部分构成,但是这2 种组分都大多来源于不可再生的石化资源,且活性稀释剂苯乙烯具有高挥发性,是一种潜在的致癌物和空气污染物。随着人们环保意识的提高以及材料可持续发展和达成碳中和目标的需求,寻求可再生资源替代石化资源作为原料制备环境友好的新型热固性高分子材料受到人们的关注,研究者近年来在生物质基不饱和聚酯基体树脂与环保型活性稀释剂两方面开展了很多研究。
在生物质基不饱和聚酯基体树脂研究方面,由于植物油具有资源丰富、可再生、成本低廉等特点,已被报道用于制备植物油基不饱和聚酯和乙烯酯基体树脂[3]。利用大豆油为原料,通过环氧化和丙烯酸化改性两步法,可以制得含有活性不饱和碳碳双键的环氧化大豆油丙烯酸酯(Acrylated epoxidized soybean oil,AESO),进一步利用马来酸酐对AESO进行酯化改性可以得到双键功能化大豆油树脂(MAESO),这2 种树脂已经实现了工业化制备。但是,大豆油是一种可食用油,作为制备材料存在与食品竞争的问题,而蓖麻油是一种重要的不可食用的工业植物油,分子结构中含有不饱和碳碳双键、羟基和酯基等反应活性基团[4]。因而,蓖麻油被视为一种替代石油资源用于制备不饱和聚酯或乙烯酯基体树脂的潜在理想原料。
在环保型活性稀释剂研究方面,已有研究利用丁香酚、异山梨醇等生物质资源来制备活性稀释剂替代苯乙烯用于植物油基不饱和聚酯。丁香酚作为一种具有刚性结构的天然生物质原料,结构中含有酚羟基和不饱和碳碳双键。酚羟基能够发生多种反应,因而已经用于制备环氧树脂、丙烯酸酯、苯并噁嗪等多种树脂[5]。Liu 等[6]以AESO 为基体树脂、甲基丙烯酸化丁香酚为活性稀释剂,通过自由基共聚反应制备了一种生物质基的大豆油基不饱和聚酯材料。这种材料具有较高的力学强度和模量,且在300 ℃时的质量损失仅5%,表现出良好的热稳定性。此外,松香[7]、腰果酚[8]、愈创木酚[9]、香草醇[10,11]、糠醛[12]、木质素[13]和蓖麻油[14]等生物质原料均可以通过双键化改性引入双键后用作AESO 或者MAESO 树脂的活性稀释剂,并制备得到性能优异的植物油基不饱和聚酯材料。本文以蓖麻油为原料,通过与甲基丙烯酸酐进行一步酯化法制备了蓖麻油甲基丙烯酸酯(MCO)基体树脂,然后以丁香酚为原料,通过酯化反应制备了甲基丙烯酸化丁香酚(ME)活性稀释剂,并将MCO 与ME 按照不同的质量比进行固化得到了蓖麻油基热固性不饱和聚酯材料,系统考察了所制备的蓖麻油基热固性不饱和聚酯材料的性能。
1 实验部分
1.1 原料
蓖麻油:购自天津科密欧化学试剂有限公司;丁香酚:购自上海麦克林生化科技有限公司;三乙胺:购自天津市富宇精细化工有限公司;甲基丙烯酸酐、4-二甲基氨基吡啶、过氧化苯甲酸叔丁酯:购自上海阿拉丁试剂有限公司;二氯甲烷、碳酸氢钠、盐酸、氢氧化钠、氯化钠和无水硫酸镁:均由天津市天力化学试剂有限公司提供。

Scheme 1 Synthesis of MCO
1.2 MCO 的合成
将10.0 g 蓖麻油、3.54 g 三乙胺和0.084 g 4-二甲氨基吡啶置于100 mL 的三口烧瓶中,并以320 r/min的转速搅拌,在保证反应装置气密性良好的条件下,持续通入氩气使体系维持惰性气氛,随后缓慢滴加4.96 g 甲基丙烯酸酐,滴完后升温至45 ℃反应24 h。将得到的粗产物用二氯甲烷进行萃取,随后用饱和NaHCO3溶液进行洗涤,直至无气泡产生;再用0.5 mol/L 的NaOH 溶 液、0.5 mol/L 的HCl 溶 液 和饱和食盐水依次进行洗涤,用分液漏斗进行油水分离,在油相中加入无水硫酸镁干燥、过滤、旋蒸即得到产物MCO,产物为淡黄色液体。
1.3 ME 的合成
称取10.0 g 丁香酚与0.16 g 4-二甲氨基吡啶于三口烧瓶中搅拌混合均匀,并持续通入氩气进行保护,然后缓慢滴加10.33 g 甲基丙烯酸酐,滴完后升温至45 ℃反应24 h。将得到的产物用二氯甲烷进行萃取,并用饱和NaHCO3洗涤至无气泡产生,再分别用1 mol/L 的NaOH 溶液、0.5 mol/L 的NaOH 溶液、0.5 mol/L 的HCl 溶液和饱和食盐水进行洗涤;洗完后分液,并在油相中加入无水硫酸镁干燥,最后过滤、旋蒸即得到产物ME,产物为浅黄色液体。
Scheme 2 Synthesis of MV
1.4 蓖麻油基不饱和聚酯的制备
以MCO 为基体树脂、ME 为活性稀释剂、过氧化苯甲酸叔丁酯为引发剂,将MCO 与ME 按照6 种不同的质量比(100:0,80:20,60:40,40:60,20:80,0:100)进行混合,并通过自由基共聚反应固化制备蓖麻油基不饱和聚酯材料。固化条件为:120 ℃固化2 h,140 ℃固化4 h,160 ℃后固化2 h,最后在烘箱中自然冷却至室温,固化后的样品如Fig.1 所示。

Fig.1 Photos of MCO-ME thermoset samples with different ME loadings
1.5 测试与表征
1.5.1 核磁共振波谱分析:使用Varian VXR-300 NMR 仪器对MCO 和ME 进行核磁氢谱表征。
1.5.2 傅里叶变换红外光谱分析:采用VECTOR-22型傅里叶变换红外光谱仪通过KBr 压片方式对MCO 树脂、ME 单体及固化后的MCO-ME 热固性材料进行红外测试,波长范围为400~4500 cm-1。
1.5.3 黏度测试:采用NDJ-8S 数字型黏度计对合成的MCO 树脂和ME 单体进行黏度测试。
1.5.4 凝胶含量测试:固化后的MCO-ME 样品的质量记为m1,然后在索氏抽提器中用二氯甲烷抽提24 h。抽提结束后,取出样品在60 °C 的烘箱烘至恒量并称量剩余样品的质量(m2),凝胶含量(W)按照式(1)计算

1.5.5 动态力学热分析:使用美国TA 公司Q800 型动态力学热分析仪(DMA)测试交联固化后MCOME 材料的动态力学性能,玻璃化转变温度选取损耗因子最大时对应的温度。所有样品(长条形,尺寸为3 mm 宽×0.25 mm 厚)按照拉伸模式以3 °C/min的速率从-50 ℃升温至180 °C 进行测试,并且维持1 Hz 的恒定频率和0.15%的应变。
1.5.6 差示扫描量热分析(DSC):在氮气气氛下,将
5~10 mg 固化后的MCO-ME 材料样品置于密闭的铝坩锅中,使用STA449F3-1053-M 型DSC 在动态扫描 模 式 下 以10 °C/min 的 升 温 速 率 从30 °C 扫 描 到220 °C,观察过程中的热量变化。
1.5.7 热重分析(TGA):MCO-ME 固化样品的热稳定性采用德国耐驰STA 449 F5 型热重分析仪进行测 试。将5~10 mg 样 品 以10 °C/min 的 扫 描 速 率 在室温至800 °C 的范围内进行热重测试,氮气气流为25 mL/min。
2 结果与讨论
2.1 MCO 和ME 的 结 构 表 征
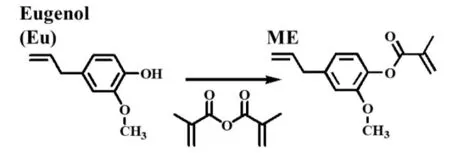
2.1.1 MCO 的结构表征:由Fig.2 中蓖麻油和MCO的核磁氢谱图可知,蓖麻油在δ5.38 和5.46 处存在不饱和碳碳双键氢(峰10 和11,—CH=CH—)的特征吸收峰,在δ3.60 处存在与醇羟基相连的叔碳氢原子的特征吸收峰(峰13,—CH—OH),在δ1.78 处存在叔碳醇羟基中氢原子的特征吸收峰(峰20,—CH—OH)。当蓖麻油与甲基丙烯酸酐反应后,蓖麻油在δ1.78 处的羟基氢吸收峰消失(峰20,—CH—OH),表明蓖麻油中的羟基发生了酯化反应,从而形成了具有吸电子能力的甲基丙烯酸酯基团。此外,产物在δ6.08 和5.53 处出现了甲基丙烯酸酯基中不饱和碳碳双键氢的特征峰(峰21,—C=CH2),且在δ1.94 处存在甲基丙烯酸酯基中甲基氢的特征吸收峰(峰22,—C—CH2),这表明蓖麻油与甲基丙烯酸酐反应成功合成了MCO。
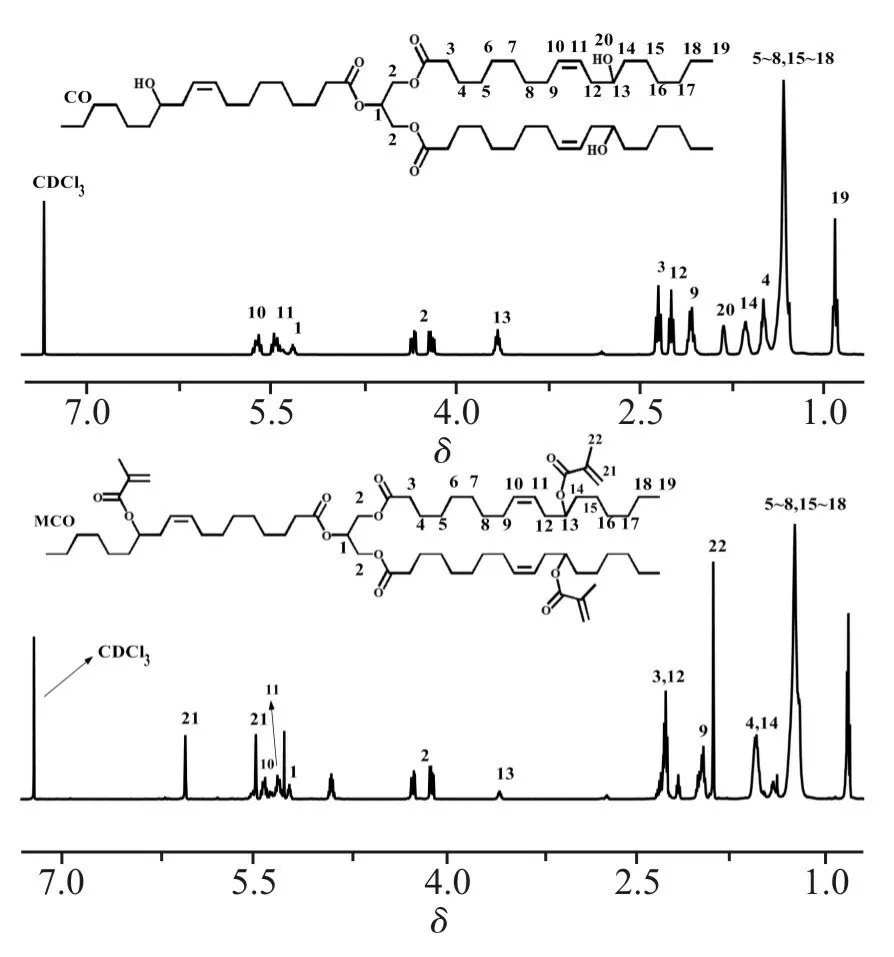
Fig.21H-NMR spectra of CO and MCO
如Fig.3 所示为CO 与MCO 的红外谱图。由图可知,CO 在3500 cm-1附近存在羟基—OH 的特征吸收峰,在1743 cm-1处存在酯基C=O 的伸缩振动吸收峰,在3005 cm-1和1640 cm-1处分别存在=C—H 和—C=C—的伸缩振动吸收峰。当CO 与甲基丙烯酸酐反应后,CO 在3500 cm-1处的—OH 伸缩振动吸收峰明显变小,在1640 cm-1处的—C=C—的伸缩振动峰增强,这些进一步表明成功制备了MCO。
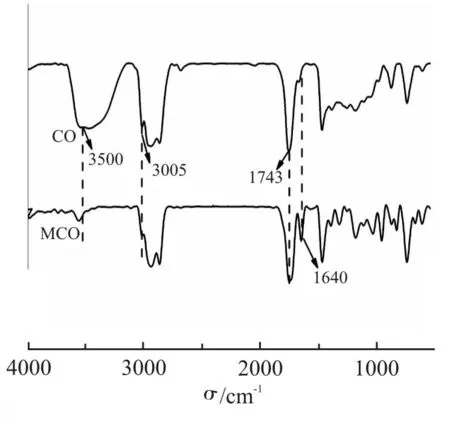
Fig.3 FT-IR spectra of CO and MCO
2.1.2 ME 的结构表征:Fig.4 为丁香酚和ME 的核磁氢谱图。由图可知,丁香酚中酚羟基(Ar—OH)的特征吸收峰位于δ8.65~8.70 处。当丁香酚与甲基丙烯酸酐反应后,位于δ8.65~8.70处的酚羟基吸收峰完全消失,而δ5.75~5.86和δ6.25~6.40(—C(CH3)=CH2),以及δ2.06~2.21(—C(CH3)=CH2)处出现了属于甲基丙烯酸酯基团的特征吸收峰,这表明成功制备了ME。

Fig.4 1H-NMR spectra of EU and ME
Fig.5 所示为丁香酚与ME 的红外谱图。如Fig.5所示,当丁香酚与甲基丙烯酸酐反应后,丁香酚在3500 cm-1处酚羟基的伸缩振动吸收峰消失,在1740 cm-1处出现了新的甲基丙烯酸酯酯键的伸缩振动峰,说明丁香酚的酚羟基充分发生酯化,成功合成了ME。
Fig.5 FT-IR spectra of EU and ME
2.2 MCO 和ME 的 黏 度
如Tab.1 所示,基体树脂MCO 在30 ℃的黏度为362.9 mPa·s,而ME在30 ℃时的黏度为13.66 mPa·s,二者以不同的质量比混合后,体系的黏度较小,满足实际的应用需求。

Tab.1 Viscosity of MCO and ME
2.3 蓖麻油基不饱和聚酯体系的固化特性
以二氯甲烷为溶剂,将固化后的蓖麻油基热固性不饱和聚酯材料进行24 h 的索氏抽提,抽提后材料的凝胶率如Fig.6 所示。由图可知,所有固化样品的凝胶率均超过91%,表明大部分MCO 和ME 都通过自由基共聚反应交联固化到三维网络结构中,材料的交联固化较为完全。固化样品中可溶于二氯甲烷的部分可能主要是MCO 和ME 聚合形成的寡聚物及少量未完全参与反应的MCO 或ME 单体的混合物。

Fig.6 Gel content of MCO-ME thermosets with different ME loadings
Fig.7 所示为不同MCO 和ME 质量比制备的固化蓖麻油基不饱和聚酯材料的DSC 曲线。由图可知,在50~180 ℃的温度范围内,所有固化后的样品都没有出现放热峰,这表明所制备的蓖麻油基不饱和聚酯材料已经完全固化,这与上述凝胶率测试结果一致。

Fig.7 DSC of cured MCO- ME thermosets with different ME loadings
2.4 蓖麻油基不饱和聚酯材料的动态力学性能
Fig.8 所示为不同MCO 和ME 质量比制备的蓖麻油基不饱和聚酯材料的储能模量和损耗因子(tanδ)随温度变化的结果。由Fig.8 (a)可知,随着温度从-50 ℃升高到200 ℃,材料的储能模量呈降低的趋势。这是因为随着温度的升高,材料中分子链段的运动能力增强,自由体积增加,使其刚性降低。纯ME 固化材料在25 ℃时的储能模量约为3090 MPa,这主要是由于ME 分子结构中含有刚性的苯环结构,因而,使得交联固化后的材料体系表现出很高的脆性,材料在固化过程中便容易发生破碎断裂。而纯MCO 材料在25 ℃时的储能模量约为4.3 MPa,约为纯ME 树脂的1/718,这主要是因为MCO分子的主链是由柔性的C—O 键和C—C 键所组成的脂肪长碳链结构,表现出很强的柔性。随着具有柔性结构的MCO 引入到刚性的ME 树脂中,所制备的材料在室温条件下的储能模量表现出降低的趋势。当MCO 与ME 的质量比为20:80 时,材料在25 ℃的储存模量最高,达1830.3 MPa。

Fig.8 DMA of MCO-ME thermosets with different ME loadings(a):storage modulus vs.temperature;(b):tanδvs.temperature
通常来说,tanδ的高度代表材料的阻尼行为,tanδ峰的高度越高,树脂体系的交联度越低;tanδ峰越低,树脂体系的交联度越高。由Fig.8 (b)可知,纯ME 固化材料的Tg高达150 ℃,而纯MCO 材料的Tg仅为-9.8 ℃,随着具有柔性结构的MCO 用量在材料体系中从20%增加到80%,MCO-ME 材料体系的玻璃化转变温度从103.6 ℃降低为-9.3 ℃。当MCO 与ME 的质量比为20:80 时,材料体系中出现2 个玻璃化转变温度峰,一个峰位于45.7 ℃,另一个峰位于108.2 ℃。这主要是因为20%的MCO 和80%的ME树脂之间的相容性不理想,当MCO-ME 树脂体系固化时,既存在MCO 树脂的自聚及ME 单体的自聚,也存在MCO 和ME 树脂的共聚,因而固化后的材料出现了富MCO 区域和富ME 区域,表现出相分离特性。而当MCO 与ME 的质量比为40:60 时,所制备的材料只有1 个玻璃化转变温度,为19.7 ℃。此外,随着ME 用量从0%增加到80%,所制备的蓖麻油基热固性材料tanδ峰的高度表现出逐渐降低的趋势,这表明随着ME 用量的增加,材料体系的交联度表现出增加的趋势。这主要是因为相同质量的ME 和MCO,ME 含有的碳碳双键数目更多。但当ME 的质量分数为80%时,材料体系出现了相分离行为,因而,当ME 的质量分数为60%时,材料体系具有最高的交联度。
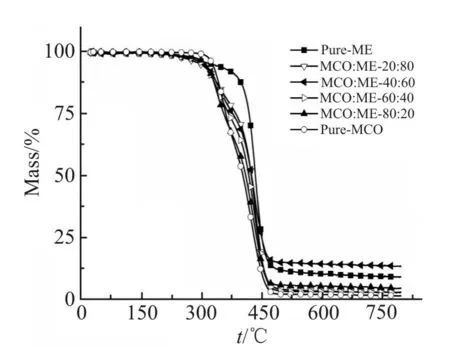
Fi g.9 TGA curves of MCO-ME thermosets with different ME loadings
不同MCO 和ME 质量比制备的蓖麻油基不饱和聚酯材料在氮气气氛下的TGA 曲线如Fig.9 所示。由图可知,所有固化样品的初始分解温度都接近320 ℃,因而样品表现出良好的热稳定性。固化的纯ME 不饱和聚酯材料在失重10%时的温度为388.1 ℃,然而,随着体系中MCO 含量的增加,材料失重10%所对应的温度有所下降。这是因为在惰性气氛中,具有刚性苯环结构的ME 比具有柔性脂肪链结构的MCO 的热稳定性更高。通常来说,残炭量与材料体系中所含苯环结构的含量及材料体系的交联度密切相关[15],相同质量下,ME 比MCO 含有更多的苯环结构,且随着ME 质量分数从0%增加到60%,材料体系的交联度表现出增加的趋势;且在ME 质量分数为60%时,材料体系的交联度达到最大(这与DMA 的测试结果一致)。因而,当MCO与ME 质量比为40:60 时,材料在800 ℃时的残炭量达到最大值13.28%。
3 结论
本文以生物质资源蓖麻油和丁香酚为原料,分别对其进行双键化改性制备得到了MCO 和ME,再将MCO 与ME 按照不同的质量比共混固化制得一系列蓖麻油基热固性不饱和聚酯材料。当m(MCO):m(ME)=40:60 时,所制备材料的综合性能最佳,玻璃化转变温度为19.7 ℃,25 ℃时材料的储能模量为288.8 MPa,凝胶率为91.4%,800 ℃时的残炭量高达13.28%,本研究对热固性不饱和聚酯材料的绿色制备提供了参考。
猜你喜欢
广州化工(2022年11期)2022-06-29
陕西科技大学学报(2022年3期)2022-05-27
科技信息·学术版(2021年4期)2021-12-30
中国水产(2021年11期)2021-12-15
粘接(2021年5期)2021-06-29
科学技术创新(2021年5期)2021-03-17
渔业研究(2020年5期)2020-10-28
森林工程(2020年5期)2020-09-17
核化学与放射化学(2020年4期)2020-08-21
中小学信息技术教育(2019年1期)2019-03-04
