
基于原位红外光谱的NiAu双金属催化剂合金化过程研究
2022-08-25 16:55张晓奔王明瑞张光辉郭新闻
天然气化工—C1化学与化工 2022年4期
张 丽,朱 杰,张晓奔,刘 伟,刘 怡,王明瑞,张光辉,郭新闻
(1.大连理工大学 化工学院,精细化工国家重点实验室,宾州-大连联合能源研究中心,辽宁 大连 116024;2.中国科学院 大连化学物理研究所 大连洁净能源国家实验室,辽宁 大连 116023)
与单金属纳米颗粒相比,双金属催化剂具有独特的催化性能,并可以通过调控活性中心的电子结构和几何结构提高其催化反应性能,因此近年来 双金属催化剂研究引起了广泛的关注[1-2]。合金化是双金属催化剂中一种普遍现象,两种金属的密切接触通常使其具有协同效应[3-4]。然而,催化剂的颗粒尺寸、双金属组成和处理条件(温度、气氛)等多种因素都可能影响合金化过程[5-7],且合金催化剂在催化反应过程中容易发生烧结、相变和表面重构等变化[8-9]。因此,理解双金属催化剂的结构演变,尤其是合金化和相分离过程,对于调控催化剂表面结构和设计高效合金催化剂具有重要意义[10-11]。
双金属催化剂的电子结构和几何结构可以通过选用不同的金属或改变金属原子比例来调控。由VIII族和IB族金属组成的双金属催化剂具有广泛的应用,如Ni-Cu[12-13]、Pd-Ag[14-15]、Pd-Au[16-17]和Cu-Au[18-19]。其中,NiAu双金属催化剂在水蒸气重整、加氢脱氯和选择性加氢等多相催化反应中表现出巨大潜力[20-21]。然而,Ni和Au在低温下不易形成体相合金。NIELSEN等[22]通过扫描透射电子显微镜(STEM)观察了Au沉积在Ni(110)晶面时的结构变化,发现在低覆盖度下金原子会取代最外层邻近的Ni原子,形成稳定的表面NiAu合金。低能离子散射与低能电子衍射结果也表明NiAu合金是在最外层而非在体相形成[23]。ZHANG等[5]通过原位电子显微镜研究了逆水煤气变换(RWGS)反应过程中NiAu双金属催化剂的结构演变,发现在高温H2下还原后,Au会迁移至Ni颗粒表面形成Ni@Au核壳结构。在RWGS反应中,内部的Ni在反应产物CO的诱导下迁移至表面,形成NiAu表面合金。
NiAu双金属催化剂在多相催化中有重要应用和潜力,但其合金化规律尚未明确,如NiAu原子比和合金化条件的影响等。本文合成不同NiAu原子比的双金属催化剂,利用CO吸附原位红外光谱(IR),结合RWGS反应性能研究其合金化过程及影响因素(包括NiAu原子比和操作条件等),并讨论该过程的热力学函数变,以期为理解NiAu双金属催化剂的合金化过程及设计开发高效NiAu双金属催化剂提供思路和参考。
1 实验部分
1.1 实验材料与试剂
正己烷(分析纯)和乙腈(分析纯)购自天津富宇精细化工有限公司;乙醇(分析纯)购自天津大茂化学试剂有限公司;油胺(OAm)(≥98%)购自Sigma-Aldrich(上海)贸易有限公司;乙酰丙酮镍(II)(Ni(acac)2,95%)、氯金酸(HAuCl4·xH2O,w(Au) ≥47.5%)和二氧化硅(60~80目)购自阿拉丁试剂公司;Ar(99.99%)、CO2(99.99%)、H2(99.99%)和N2(99.99%)均购自大连化物所气体有限公司。所有试剂使用时均未经进一步纯化。
1.2 催化剂制备
NiAu/SiO2催化剂采用前期报道的方法制备[5]。将40 mL OAm加入到三口烧瓶中,使用双排管将三口烧瓶中的空气置换成高纯Ar,并使烧瓶内始终处于惰性气氛。在搅拌状态下加热至100 °C并保持20 min。加入2 g Ni(acac)2搅拌10 min后形成透明的蓝绿色溶液,再次用双排管除去加药品带进的少量空气。在持续搅拌下将10 mL HAuCl4的乙醇溶液(0.02 g/mL)缓慢滴加入上述溶液中,在100 °C下保持60 min至溶液变为紫黑色,快速升温至230 °C继续反应90 min,然后在Ar气氛下自然降温。用正己烷和乙醇的混合溶液(V正己烷:V乙醇=3:1)离心洗涤产物,将获得的NiAu纳米颗粒分散在乙醇中并与一定质量的二氧化硅混合,在45 °C下旋蒸除去乙醇。将负载后的催化剂用乙腈在85 °C下冷凝回流10 h除去残留有机物,最后在60 °C下过夜干燥,200 °C下焙烧4 h。通过改变前驱体Ni(acac)2的量,使用相同的方法制备不同NiAu原子比的NiAu/SiO2催化剂。催化剂命名为NiAu-x,其中x代表NiAu原子比。
1.3 催化反应性能评价
RWGS反应在固定床反应器中进行。取0.2 g催化剂装填于直径为8 mm的石英管中,在550 °C、常压下用纯H2(50 mL/min)还原90 min,然后切换至反应气(V(CO2):V(H2):V(N2) =1:3:4),流速为40 mL/min。产物使用在线气相色谱(安捷伦7890B)分析。CO2的转化率(X,%)、产物的选择性(S,%)的计算公式如下:
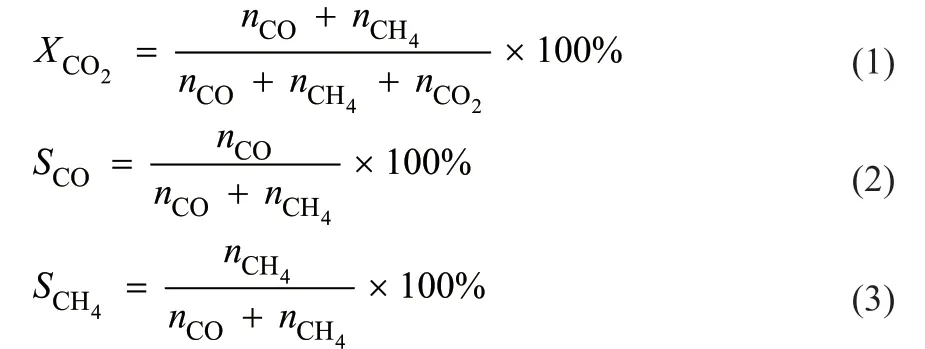
1.4 实验表征
X射线粉末衍射(XRD)谱图在日本Rigaku公司的SmartLab 9 kW衍射仪上采集,采用Cu Kα(λ=0.15406 nm)射线,管电压为45 kV、管电流为200 mA,扫描角度2θ的范围为10°至80°。
透射电子显微镜(TEM)图像在HT7700 EXALENS(日本日立公司)仪器上采集,加速电压为120 kV;高分辨透射电子显微镜(HRTEM)图像在JEM F200(日本电子(JEOL)公司)仪器上采集,加速电压为200 kV,能谱(EDS)分析在扫描透射模式下进行;高角度环形暗场扫描透射电子显微镜(HAADF-STEM)图像在Themis-ETEM仪器(美国Thermo Scientific公司)上采集,加速电压为300 kV。首先将样品分散在乙醇中,超声处理10 min,然后将悬浮液滴在铜网格上进行成像。
电感耦合等离子体原子发射光谱(ICP-AES)用于测定Ni和Au的实际负载量,使用PerkinElmer AVIO 500(美国PerkinElmer公司)仪器进行分析。
原位漫反射傅里叶变换红外(DRIFT)实验在Thermo Scientific Nicolet iS50(美国Thermo Scientific公司)光谱仪上进行。将粉末样品装入配有ZnSe窗口的高温原位池中,使用碲化镉汞(MCT)检测器采集光谱,分辨率为8 cm-1。对于CO吸附原位红外(CO-DRIFT)实验,先将样品在550 °C、常压下用H2/N2(φ(H2) =80%)还原30 min,然后冷却至室温以采集背景光谱。再通入CO/N2(φ(CO) =10%)10 min后,用N2吹扫后采集红外光谱。对于RWGS反应过程中的原位实验,背景光谱在550 °C的H2预还原条件下采集,之后在反应气(V(CO2):V(H2) =1:3)中550 °C下连续采集光谱。
2 结果与讨论
2.1 催化剂结构性质
NiAu-x的晶体结构通过XRD表征得出,结果如图1所示。44.4°的衍射峰对应Ni(111)晶面衍射。NiAu-x催化剂中没有观察到Au的衍射峰,表明Au是高分散的。随着NiAu原子比降低,Ni(111)晶面的衍射峰变弱。未负载的单分散NiAu纳米颗粒的形貌如图2(a)~(d)所示,Ni与Au紧密连接,形成双元纺锤形纳米颗粒。图2(b)中的选区电子衍射(SAED)结果与金属Ni和Au相对应,与XRD结果相符。如图2(e)~(j)所示,NiAu纳米颗粒NiAu-4、NiAu-8和NiAu-16的平均粒径分别为15 nm、12 nm和25 nm,且均匀分布在载体二氧化硅表面。颗粒的均匀分散以及Ni与Au的紧密接触为后续合金化研究提供了基础。表1是NiAu-x的元素分析(ICP-AES)结果,实际NiAu原子比略高于投料比,实际值与预测值基本吻合。

表1 NiAu-x和Au催化剂的ICP-AES结果Table 1 ICP-AES results of NiAu-x and Au catalysts
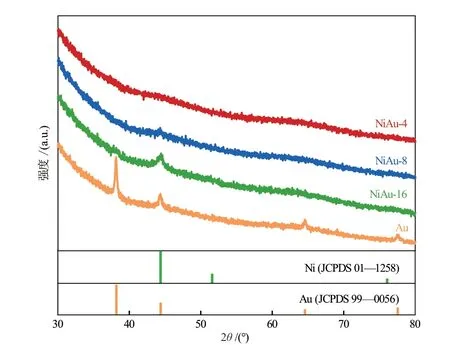
图1 新制备的NiAu-x和Au催化剂的XRD谱图Fig.1 XRD patterns of fresh NiAu-x and Au catalysts
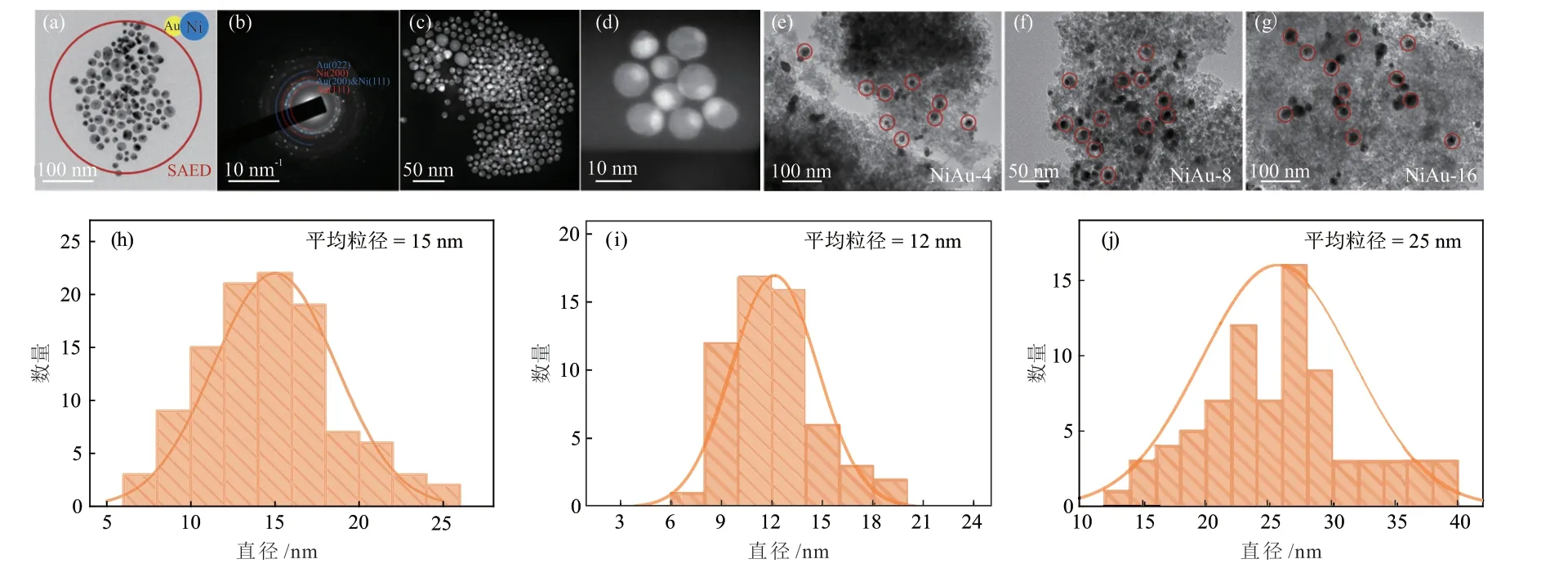
图2 NiAu-x催化剂的电镜分析及粒径分析结果Fig.2 Electron microscopic analysis and particle size analysis results of NiAu-x catalysts
2.2 催化剂的表面合金化
为了研究NiAu的合金化过程,本文采用CODRIFT实验跟踪NiAu-x催化剂表面结构的变化,如图3所示(图中标注的时间是指室温饱和吸附CO后N2吹扫的时间)。如图3(a)所示,550 °C还原后的NiAu-4在2100 cm-1和2051 cm-1处出现CO吸附峰,分别归属于CO在金和镍原子上的吸附[24-25]。前期研究表明,RWGS反应可以诱导表面NiAu合金形成[5]。进一步对RWGS反应后NiAu-4上的CO吸附进行研究,结果如图3(a)所示:Au位点上的CO吸附峰(Au-CO)向低波数移动至2088 cm-1。如图3(b)~(c)所示,NiAu-8和NiAu-16上也存在类似变化,CO吸附峰分别从2093 cm-1、2089 cm-1偏移至2085 cm-1、2083 cm-1。相关研究表明,Au与其它过渡金属合金化时,双金属的电子效应通常会使CO吸附峰发生偏移。如WEI等[6]利用CO吸附原位红外发现在PdAu合金中,Pd向Au电子转移使Pd上的CO吸附峰向高波数偏移。AuCu合金的CO吸附原位红外表明,随AuCu原子比增大,Cu向Au转移电子增加,因此Cu上的CO吸附峰逐渐向高波数偏移[26]。前期研究也观察到[5],相比于CO在Au上的吸附,其在NiAu合金上的吸附峰向低波数偏移。本研究中,不同NiAu原子比的催化剂在RWGS反应后Au上的CO的吸附峰均向低波数偏移,表明NiAu合金形成。在合金化过程中几乎未观察到Ni位点上CO吸附峰(Ni-CO)的变化,可能是由于催化剂中Ni的含量都远高于Au,形成的Ni—Au键远少于Ni—Ni键,即纳米颗粒表面镍原子的周围与之成键的仍然多数为镍原子。
Ni—Au键的键能(0.80 eV×2)低于Ni—Ni键(1.03 eV)和Au—Au键(0.59 eV)的键能总和[27],NiAu合金化为吸热过程,即焓变(ΔH)大于0,同时合金化过程熵增(ΔS>0)。根据方程ΔG=ΔH-TΔS可知,高温更有利于合金化。随着NiAu原子比增大,金原子周围近邻的镍原子数目增加,形成合金需要断裂的Ni—Ni和Au—Au键的数量减少,导致合金化的ΔH降低,更易形成合金。红外光谱结果(图3)表明,在550 °C下H2还原后,NiAu-4的Au-CO红外振动吸收出现在2100 cm-1,而NiAu原子比较高的催化剂,其Au-CO的红外振动吸收波数较低(NiAu-8为2093 cm-1,NiAu-16为2089 cm-1)。NiAu-16的Au-CO红外振动吸收波数与NiAu合金接近(2083~2088 cm-1),表明NiAu-16在550 °C H2还原后即形成合金。
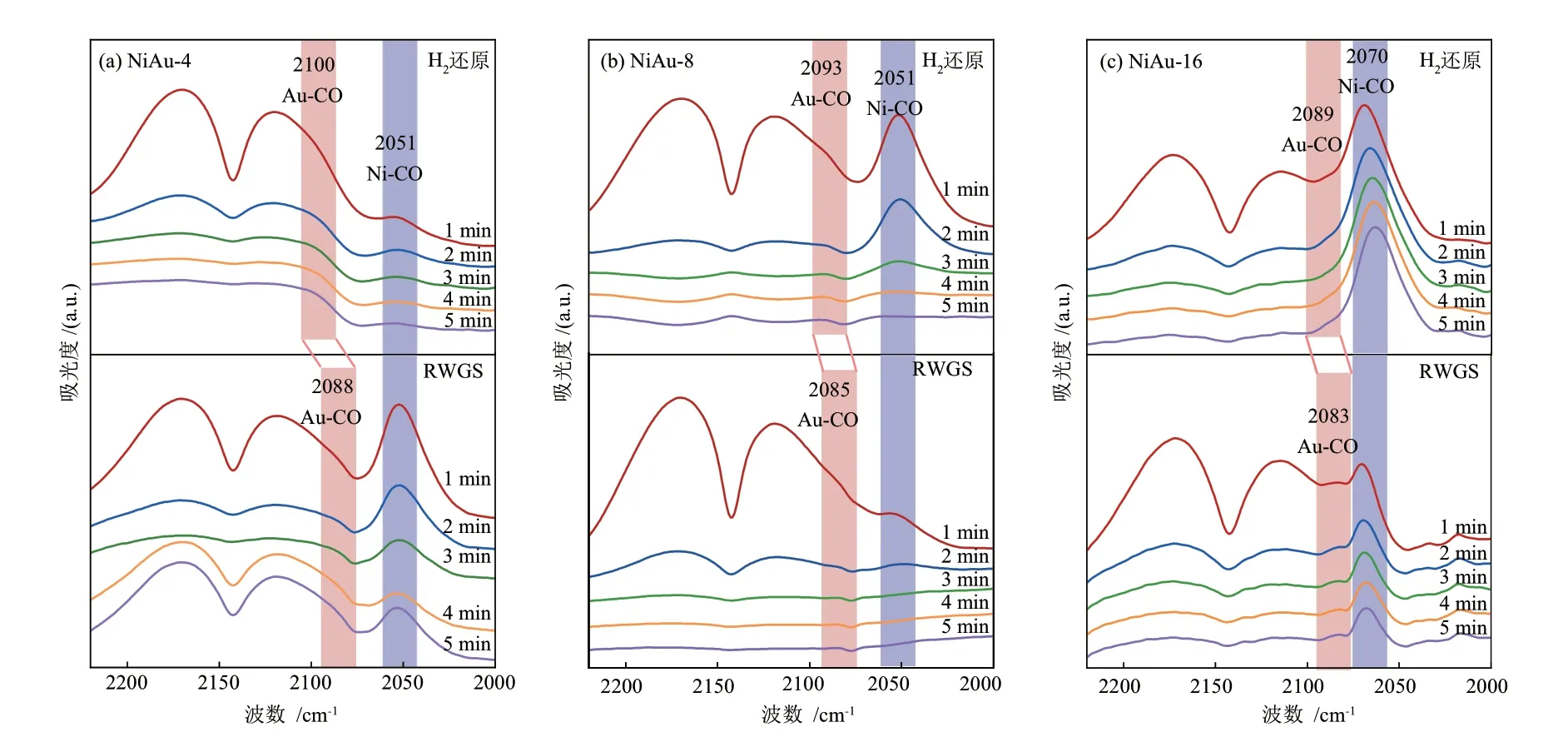
图3 NiAu-x催化剂的CO-DRIFT谱图Fig.3 CO-DRIFT spectra for NiAu-x catalysts
NiAu-4和NiAu-8催化剂在更高温度(650 °C) 下H2处理后的CO-DRIFT光谱如图4所示(图中标注的时间是指室温饱和吸附CO后N2吹扫的时间)。在2080 cm-1处均可观察到NiAu合金的Au-CO的振动吸收,表明两种催化剂均形成了NiAu合 金。NiAu-4在2095 cm-1处 仍 存 在 未 形成合金的Au-CO的振动吸收,表明在该条件下NiAu-4表面部分形成合金;与之相比,NiAu-8合金化程度更高;而NiAu-16在550 °C还原后已形成合金。这表明,高NiAu原子比时合金化的ΔH较低,需要的温度低,与前面热力学函数变的分析一致。

图4 650 °C H2还原后NiAu-x催化剂的CO-DRIFT谱图Fig.4 CO-DRIFT spectra for NiAu-x catalysts after H2reduction at 650 °C
图3的原位红外实验结果也表明,RWGS反应可以促进NiAu合金化过程[5]。反应产物CO吸附在过渡金属表面(包括Ni和Au)为放热过程,即ΔH<0。根据热力学分析,在RWGS反应中,CO在金属表面的吸附可以降低整体合金化过程的ΔH和ΔG,而H2可以维持Ni和Au的金属态,因此RWGS反应可以促进NiAu合金形成。
2.3 催化剂反应性能
RWGS反应性能也可以作为表征金属催化剂表面结构的探针。WU等[28]发现约9 nm的Ni纳米颗粒可选择性地将CO2转化为CH4,而Ni团簇表现出相对较高的CO选择性:10%的CO2转化率下,CO选择性为40%。经过H2预还原后,NiAu-x催化剂的CO2加氢性能如图5所示。
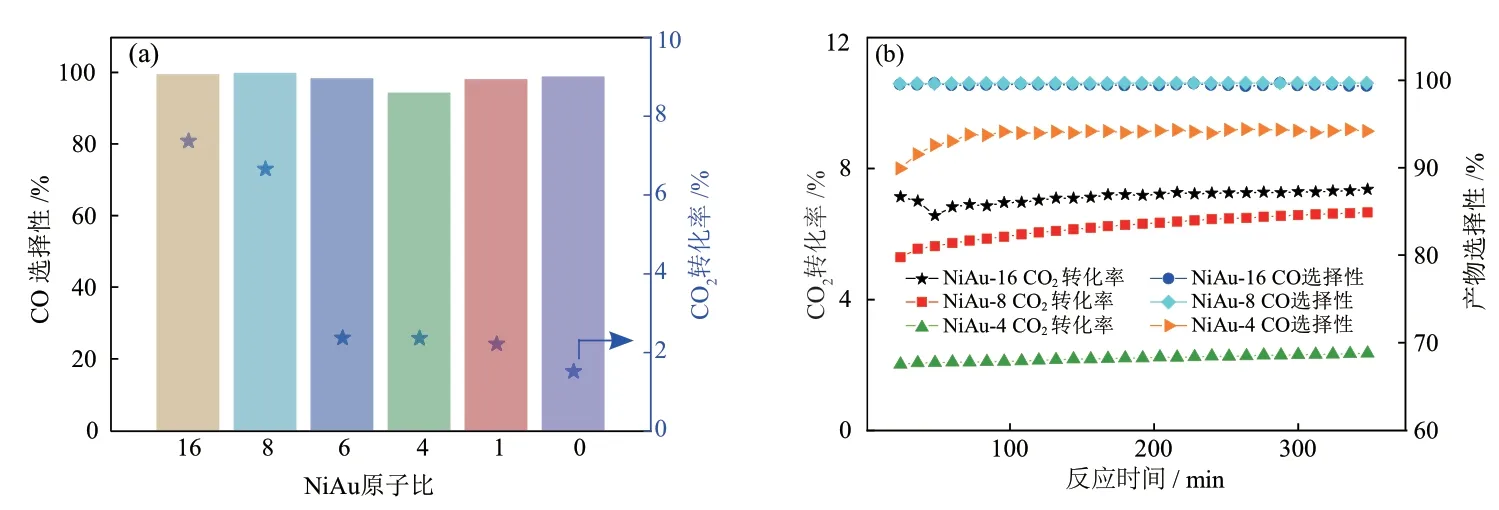
图5 NiAu-x催化剂的CO2加氢性能Fig.5 CO2hydrogenation performance of NiAu-x catalysts
如图5(a)所示,单金属Au在RWGS反应中几乎不具有催化活性,而Ni的加入提高了CO2转化率。在不同的NiAu原子比时,CO选择性始终保持95%以上,这与单原子Ni在RWGS反应中的选择性接近[29]。值得注意的是,NiAu-x催化剂的粒径均大于10 nm,高CO选择性表明其表面的镍原子被金原子阻隔,即形成NiAu表面合金。由图5(b)可以看出,在300 min内,CO2转化率和CO选择性都随反应时间(TOS)延长而升高,这与前述RWGS反应可以诱导表面NiAu合金的形成、提高CO产率的结论一致。此外,原位红外跟踪NiAu-x催化剂上RWGS反应的结果如图6所示。催化剂上均观察到气态CH4(3016 cm-1)和CO(2180 cm-1和2114 cm-1)的红外吸收峰[30-31],且CO的峰面积远高于CH4,这与其具有高CO选择性一致。

图6 RWGS反应后NiAu-x催化剂的原位红外谱图Fig.6 In situ IR spectra of NiAu-x catalysts after RWGS reaction
2.4 反应后催化剂结构表征
反应后NiAu-x催化剂的能谱分析(EDS)如图7所示。3种催化剂中Au与Ni都均匀分散,未观察到孤立的Ni和Au颗粒,与RWGS反应后形成NiAu合金的结论一致。

图7 RWGS反应后NiAu-x催化剂的EDS谱图Fig.7 EDS mappings of NiAu-x catalysts after RWGS reaction
3 结论
本文利用原位红外光谱跟踪了NiAu双金属催化剂的合金化过程,发现在一定的NiAu原子比范围内(4~16),高NiAu原子比有利于合金化。NiAu-16 在550 °C H2还原后即可形成合金,NiAu-8需在650 °C还原后形成合金,NiAu-4在650 °C后仅部分形成合金。同时,RWGS反应可以诱导NiAu合金化,NiAu-8和Ni-16在550 °C还原后经过RWGS反应可形成NiAu合金。根据热力学分析,增大NiAu原子比,合金化过程的ΔH降低;RWGS反应的产物CO在NiAu表面吸附放热,也可降低合金化过程的ΔH。因此,增大NiAu原子比、提高还原温度和RWGS反应均可降低NiAu合金化的ΔG,促进合金化过程。本工作为NiAu双金属催化剂合金化过程提供了基于原位红外光谱的实验结果和理论分析,有助于设计开发高效NiAu催化剂。
猜你喜欢
化工管理(2022年13期)2022-12-02
辽宁化工(2022年9期)2022-09-29
实用手外科杂志(2022年2期)2022-08-31
机械工程材料(2022年8期)2022-08-29
中国钱币(2022年1期)2022-08-23
中国金属通报(2022年6期)2022-06-22
陶瓷学报(2021年5期)2021-11-22
粉末冶金技术(2021年1期)2021-03-29
航天工业管理(2020年9期)2020-12-28
太空探索(2016年6期)2016-07-10
