
等离子体辅助制备低温高活性CO2催化还原用Y2O3-Ni/CeO2催化剂
2022-08-25 16:55李露明高新华
天然气化工—C1化学与化工 2022年4期
梅 加,马 军,肖 鑫,李露明,高新华,储 伟
(1.四川大学 化学工程学院,四川 成都 610065;2.成都大学 高等研究院,四川 成都 610106;3.宁夏大学 省部共建煤炭高效利用与绿色化工国家重点实验室,宁夏 银川 750021)
CO2捕集、利用和储存(CCUS)是实现“双碳”目标的一个可行和有效方法[1]。从CO2治理技术难易程度、减排效果和目前工艺的开发程度等因素综合分析,CO2催化还原制合成天然气(SNG)是CO2利用中最有应用前景的方法之一[2-3]。由于该反应受到较大的动力学限制,并且在高温下存在逆水汽变换反应、甲烷重整等竞争反应[4],因此开发一种低温高效的CO2催化还原制合成天然气的催化剂是该工艺中最为重要和关键的研究课题之一。
Ni基催化剂由于成本低廉,简单易得而常被视为Rh基[5]、Ru基[6]等贵金属催化剂的良好替代品。郭琳等[7]在整体式纳米碳负载的Ni基催化剂上加入CeO2助剂,显著提升了该催化剂的CO2催化还原反应性能。SUN等[8-9]报道了Y助剂通过增强Ni物种的还原性能和催化剂上弱碱性和中等碱性位点有效提高Ni基催化剂的CO2催化反应活性。郭芳等[10]通过等离子体辅助制备和La2O3助剂促进协同策略显著提升了CO2催化还原反应性能。然而Ni基催化剂的低温CO2活化能力有待进一步提高,并且如何解决Ni基催化剂因烧结和积炭而迅速失活的问题也成为研究关键[11]。
由于具有极高的化学反应活性的非平衡态的等离子体物种可在低温下产生,且在催化剂制备中能有效增强与等离子体物种接触的催化剂表面的反应活性。因此,等离子体辅助制备与传统的热处理方法相比,显示出优越的特性。本文研究等离子体辅助制备下Y2O3促进的Ni/CeO2催化剂上CO2催化还原制合成天然气的反应性能,考察Y2O3的含量(质量分数,下同)对催化剂低温反应活性的影响,并通过XRD、XPS、TEM、H2-TPR、CO2-TPD和H2脉冲吸附等表征方法研究催化剂结构和反应活性的关系,以期阐明催化剂优良低温反应活性的原因。
1 实验部分
1.1 实验试剂
Ni(NO3)2·6H2O(分析纯,成都市科隆化工试剂厂);CeO2(分析纯,阿拉丁试剂有限公司);Y(NO3)3·6H2O(分析纯,成都市科隆化工试剂厂)。
1.2 催化剂制备
实验采用浸渍法和N2辉光等离子体辅助处理制备了一系列不同质量分数(0、0.5%、1.5%、3.0%和5.0%)的Y2O3促进的Ni/CeO2催化剂。首先按计量称量所需质量的Ni(NO3)2·6H2O和Y(NO3)3·6H2O,在室温下用去离子水搅拌溶解。然后按计量加入CeO2,搅拌30 min至混合均匀,在室温下静置老化3 h后在水浴(80 °C)中搅拌蒸干样品,直至无液体残留,然后将搅拌蒸干后的样品置于鼓风干燥箱中,在60 °C下干燥12 h。最后将上述样品放入以N2作为生成气的辉光等离子体中,在80 W的处理功率下处理60 min,通过该方法制备了xY2O3-Ni/CeO2催化剂,其中Ni的负载量(质量分数)为10.0%,x(%)代表Y2O3的质量分数。
1.3 催化剂性能考察及稳定性测试
CO2催化还原制合成天然气反应在常压连续固定床流动反应器中进行。200 mg催化剂在40 mL/min H2中600 ℃原位还原60 min。还原结束后在Ar中冷却至反应温度,将50 mL/min混合气体(n(H2):n(CO2) =4:1)引入固定床反应器,反应气体空速(GHSV)为15000 mL/(g·h),反应温度为180~300 °C。在250 °C的恒定温度下进行80 h稳定性测试,使用在线SP-7890气相色谱仪对反应产物进行分析。CO2的转化率(XCO2,%)、CH4的选择性(SCH4,%)和生成速率(FCH4,μmol/(s·g))按照以下公式计算。
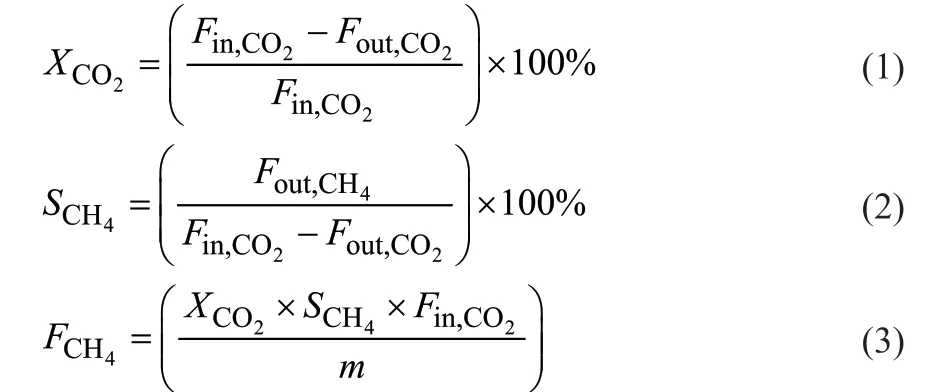
式中,Fin,CO2为CO2进气量,mL/min;Fout,CO2为CO2出气量,mL/min;Fout,CH4为CH4出气量,mL/min;m为催化剂质量,g。
1.4 催化剂表征
采用Panalytical Empyrean进行X射线衍射(XRD)测试,以λ=1.5406×10-10m的Cu Kα射线为放射源,在管电压45 kV,管电流40 mA条件下进行了XRD谱图分析。扫描范围为10°~80°,扫描速率为2 (°)/min。
采用配备了Al Kα(hv=1486.6 eV)X射线源的XSAM800光谱仪来获得X射线光电子能谱(XPS)数据,以C 1s的284.8 eV为标准校正能量。
采用FEI Tecnai G2 F20进行透射电子显微电镜(TEM)实验,加速电压为300 kV。
采用TP-5080全自动多用化学吸附仪(天津先权)进行H2程序升温还原(H2-TPR)实验。测试前,30 mg样品在400 °C、流速为50 mL/min的Ar中吹扫60 min,去除水分和吸附的杂质。反应器冷却至50 °C后,通入H2体积分数为5% H2/N2混合气,流速为30 mL/min。测试温度以10 °C/min的速率从50 ℃升温至800 °C,采用热导检测器(TCD)在线分析H2消耗。
采用与H2-TPR相同的全自动多用化学吸附仪进行CO2程序升温脱附(CO2-TPD)表征。100 mg还原后样品,在Ar气氛中降温到60 °C,完成CO2吸附后,再用Ar去除物理吸附的CO2,然后以10 °C/min的升温速率从60 °C升温至700 °C进行CO2程序升温脱附,用TCD检测器检测CO2脱附信号。
采用AUTO CHEM 2920进行H2脉冲吸附实验,100 mg样品在H2气氛中600 °C还原处理60 min后,降温至50 ℃,用高纯He吹扫多余H2待基线稳定,再用H2脉冲测试金属的分散度和粒径。
2 结果与讨论
2.1 催化剂的CO2催化还原性能评价结果
xY2O3-Ni/CeO2催化剂在CO2催化还原合成天然气反应中CO2转化率与温度的关系如图1(a)所示。随着反应温度的升高,CO2转化率升高,并且所有催化剂在测试区间内的CH4选择性都为100%。Y2O3的加入能显著提升催化剂的低温活性,在220 °C时,1.5Y2O3-Ni/CeO2催化剂具有最高的CO2转化率(84.1%),说明Y2O3的最佳负载量为1.5%,此时其具有最优异的低温反应活性。同时对比了Ni/CeO2与1.5Y2O3-Ni/CeO2在220~260 °C下的CH4生成速率,如图1(b)所示。1.5Y2O3-Ni/CeO2催化剂上的CH4生成速率均大于31.0 μmol/(g·s),远高于Ni/CeO2上CH4的生成速率,尤其在220 °C的相对低温下,1.5Y2O3-Ni/CeO2上的CH4生成速率约是Ni/CeO2的8.2倍。同时比较了文献[12-18]中报道的在温和条件(相对低温和常压)下用于CO2催化还原制合成天然气反应的Ni基催化剂的反应活性,如表1所示。
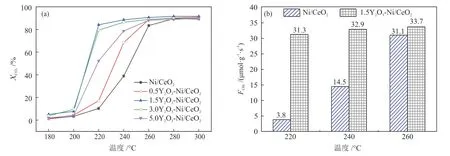
图1 xY2O3-Ni/CeO2的CO2转化率与温度的关系(a)和Ni/CeO2与1.5Y2O3-Ni/CeO2的CH4生成速率(b)Fig.1 Relationships between CO2conversion and temperature of xY2O3-Ni/CeO2catalysts (a) and CH4formation rate of Ni/CeO2and 1.5Y2O3-Ni/CeO2(b)
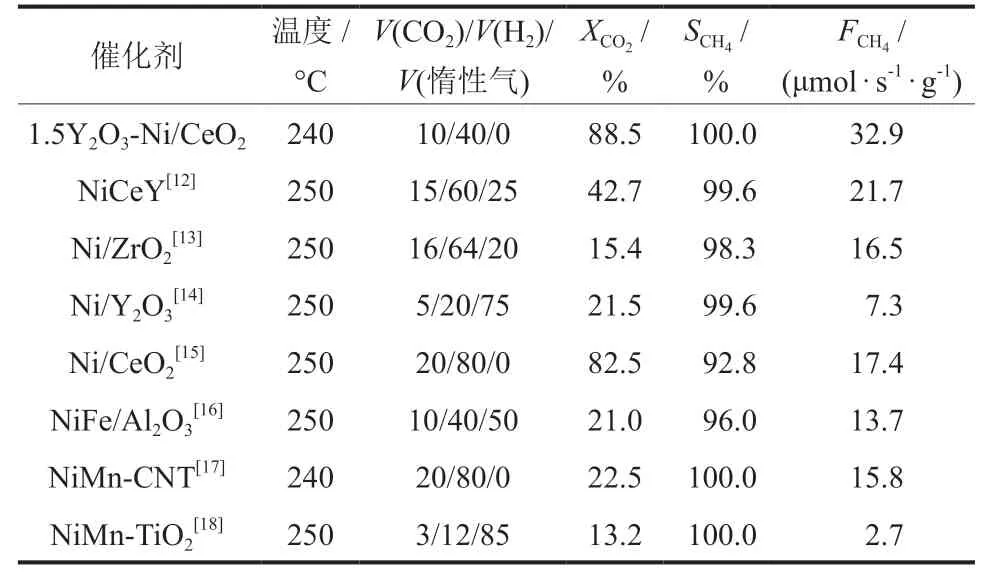
表1 温和条件下用于CO2催化还原制合成天然气反应的Ni基催化剂反应活性比较Table 1 Comparison of Ni-based catalysts for CO2catalytic reduction to SNG reaction under mild conditions
据报道[11,15],Ni基催化剂的失活通常是由活性物种的聚集和高温下的碳沉积引起的。在250 °C反应条件下,这种负面效应可以得到缓解。如图2所示,在80 h稳定性测试中,1.5Y2O3-Ni/CeO2始终保持约85.0%的CO2转化率和100%的CH4选择性,显示出良好的低温催化活性和优异的稳定性。
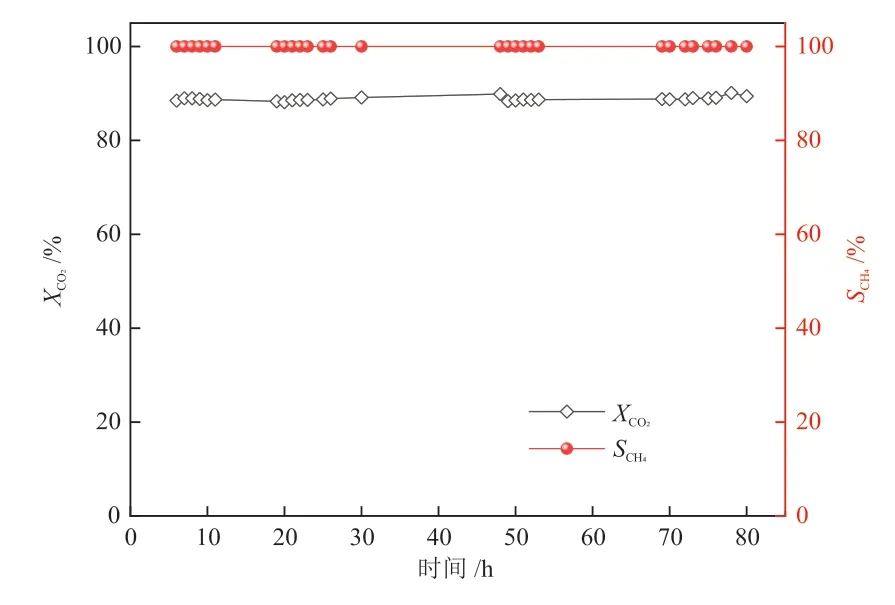
图2 1.5Y2O3-Ni/CeO2在250 °C下的稳定性测试Fig.2 Stability test of 1.5Y2O3-Ni/CeO2at 250 °C
根据Arrhenius公式,以CO2转化率的对数和温度的倒数作图,通过斜率可算出反应的表观活化能[10],如图3所示。排除内外扩散效应的影响,活化能测试过程中CO2的转化率都低于10%。Ni/CeO2的活化能为102.7 kJ/mol,而1.5Y2O3-Ni/CeO2的活化能为78.8 kJ/mol,表明Y2O3的加入能够降低反应能垒,从而加快反应速率。
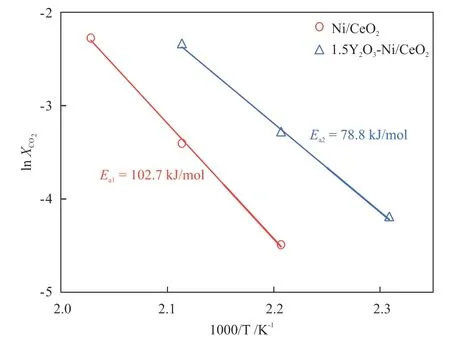
图3 Ni/CeO2与1.5Y2O3-Ni/CeO2的Arrhenius 曲线Fig.3 Arrhenius plots of Ni/CeO2and 1.5Y2O3-Ni/CeO2
2.2 催化剂的晶相结构分析及H2脉冲吸附分析
为了研究Y2O3加入对催化剂中CeO2和Ni晶体结构的影响,通过XRD研究了1.5Y2O3-Ni/CeO2和 Ni/CeO2的晶体结构,如图4所示。
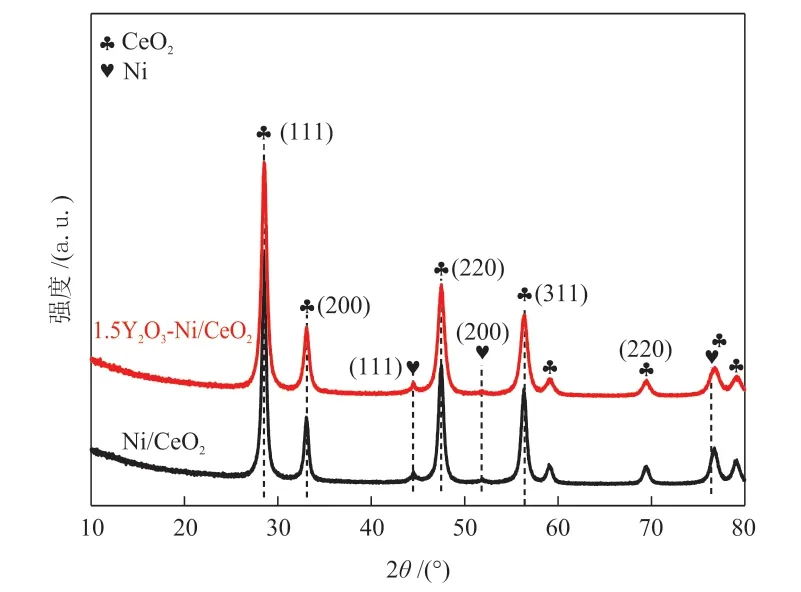
图4 还原后1.5Y2O3-Ni/CeO2和Ni/CeO2的XRD谱图Fig.4 XRD patterns of reduced 1.5Y2O3-Ni/CeO2and Ni/CeO2
两种催化剂上都检测到CeO2和Ni的特征衍射峰,但在1.5Y2O3-Ni/CeO2上未能观察到明显的Y2O3物种的特征衍射峰,这可能是由于Y2O3含量较低或结晶度较差。XRD谱图中位于2θ=28.5°、33.1°、47.5°和56.3°的4个衍射峰分别对应CeO2的(111)、(200)、(220)和(311)晶面;位于2θ=44.5°、51.8°和76.4°的特征衍射峰分别对应于Ni的(111)、(200)和(122)晶面。与Ni/CeO2相比,1.5Y2O3-Ni/CeO2的CeO2的衍射峰具有更大的半高宽。通过Scherrer公式估算的CeO2(111)的粒径以及通过H2脉冲吸附实验测得的Ni的粒径和分散度如表2所示。可以看出,1.5Y2O3-Ni/CeO2中CeO2的粒径比Ni/CeO2减小了18.7%,而Ni的粒径减小了34.2%,且Ni的分散度显著增加,表明Y2O3的加入增加了催化剂中活性位数量。
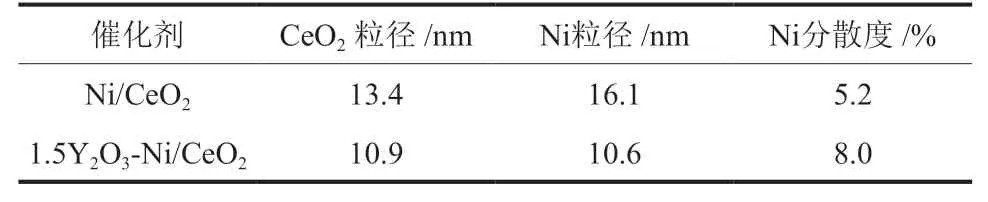
表2 还原后1.5Y2O3-Ni/CeO2和Ni/CeO2中CeO2和Ni粒径及Ni分散度Table 2 Particle sizes of CeO2and Ni and dispersion of Ni in reduced 1.5Y2O3-Ni/CeO2and Ni/CeO2
2.3 催化剂微观结构和表面组分状态分析
Ni/CeO2和1.5Y2O3-Ni/CeO2的TEM表征结果如图5所示。从图5(a)和图5(e)中可以看出,两种催化剂中CeO2均以纳米颗粒形式存在,但很难确定其粒径分布,因此为了研究催化剂表面物种的纳米结构,进行了晶格条纹分析。在Ni/CeO2上发现了晶面间距为0.201 nm和0.311 nm的晶体结构,分别属于Ni(111)和CeO2(111)纳米颗粒。在1.5Y2O3-Ni/CeO2上发现了晶面间距为0.200 nm、0.310 nm和0.304 nm 的晶体结构,分别属于Ni(111)、CeO2(111)和Y2O3(222)纳米颗粒,且CeO2和Y2O3发生了明显的晶格掺杂(图5(f))。
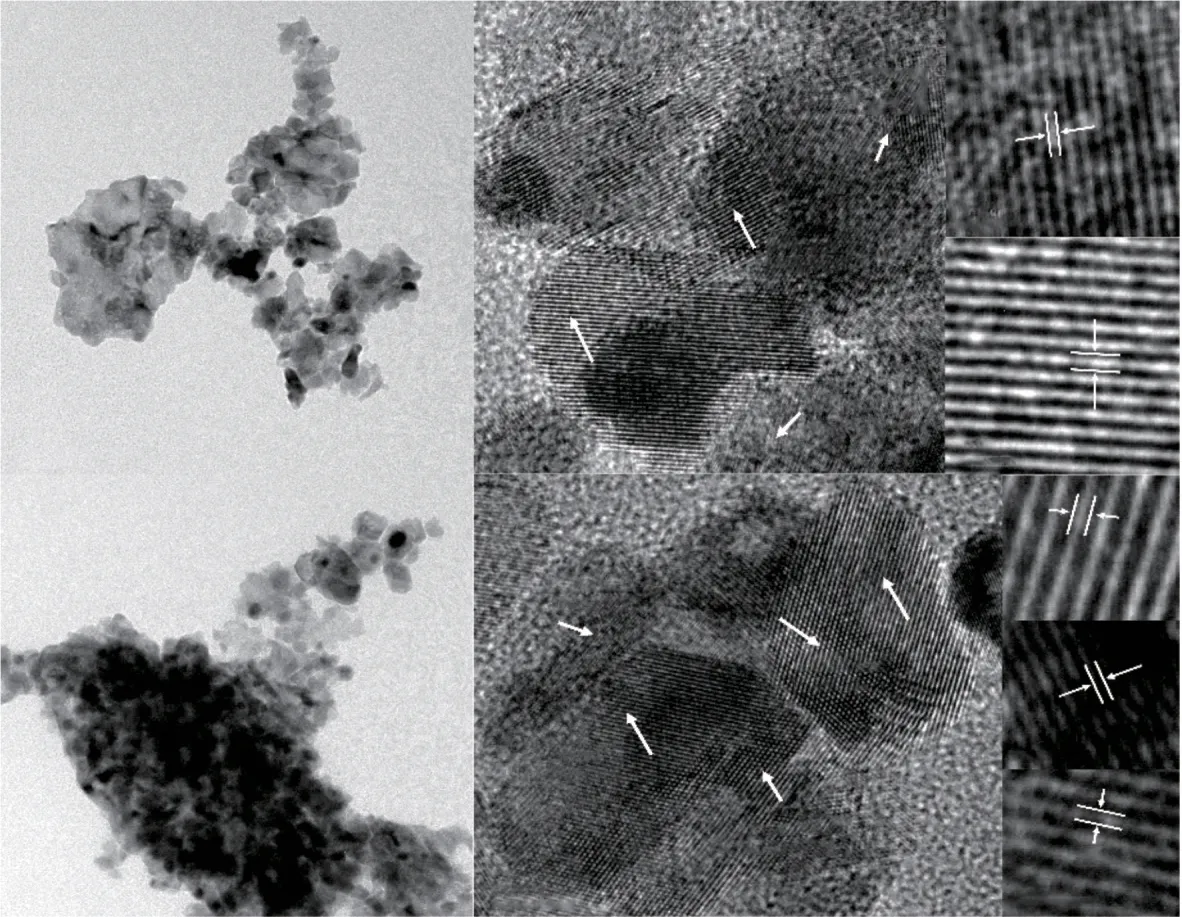
图5 还原后Ni/CeO2((a)~(d))和1.5Y2O3-Ni/CeO2((e)~(i))的TEM照片Fig.5 TEM photos of reduced Ni/CeO2((a)~(d)) and 1.5Y2O3-Ni/CeO2((e)~(i))
为揭示反应状态下催化剂表面的组分状态,对还原后的催化剂进行了XPS表征分析。催化剂的Ce 3d XPS光谱如图6(a)所示,可以分为4组峰,其中α峰(α1~α4)和β峰(β1~β4)分别对应Ce 3d3/2和Ce 3d5/2的自旋轨道,α2和β2峰对应Ce3+,而其他峰对应Ce4+物种[20]。Ni 2p3/2的XPS光谱如图6(b)所示,位于约853.0 eV处的峰归属于Ni,而约855.1 eV和约861.0 eV处分别是Ni2+及其卫星峰(NiO或者与界面处CeO2形成Ni—O—Ce相互作用的Ni2+物种)[21]。如图6(c)所示,含Y2O3的4种催化剂中,代表Y3+物种的Y 3d双峰分别位于约157.1 eV(Y 3d5/2)和约159.1 eV(Y 3d3/2),而Ni/CeO2样品中则未发现这两个特征峰。如图6(d)所示,O 1s光谱中检测到两种O物种,分别是位于约529.2 eV处的表面晶格氧(OL)和位于约531.5 eV处的表面吸附氧(OA)[22-23]。
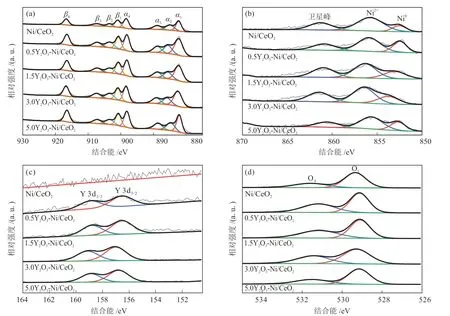
图6 还原后xY2O3-Ni/CeO2的Ce 3d (a)、Ni 2p (b)、Y 3d (c)和O 1S (d) XPS谱图Fig.6 XPS spectra of Ce 3d (a),Ni 2p (b),Y 3d (c) and O 1S (d) of reduced xY2O3-Ni/CeO2
根据XPS谱图分峰拟合的面积进行估算得到Ce3+和Ni0的相对含量以及OL与OA的比值(nOL/nOA)如表3所示。结果发现5种催化剂的Ce3+相对含量相差不大,约为20%,且呈现先减少后增加的趋势,说明Ce3+的相对含量不是催化剂性能提升的关键因素,其中1.5Y2O3-Ni/CeO2的Ce3+相对含量最小,为18.9%,但是1.5Y2O3-Ni/CeO2中Ni0的相对含量是Ni/CeO2的1.7倍,为33.7%。5种催化剂中nOL/nOA的变化呈现先减少后增加的趋势。结合TEM结果,这可能是由于等离子处理过程会产生丰富的Ni—O—Ce界面结构,并锚定小颗粒的Ni形成H2的活化中心[15,24],但是由于Y2O3的加入削弱了Ce—O键,进而促进了氧空位的产生[12],这也弥补了因Ni还原减少的部分氧空位数量。并且随Y2O3含量的增加,催化剂中氧空位含量也逐渐增加,OL占比也随之增加。

表3 表面组分相对含量和nOL/nOATable 3 Relative content of surface components and nOL/nOA
综上,在等离子体处理的催化剂中加入Y2O3,大量Ni被还原,促进了氧空位的形成。
2.4 催化剂的还原特性分析
通过H2-TPR测试了xY2O3-Ni/CeO2的还原性能,如图7所示。H2-TPR的谱峰可以分成3个峰,第1个峰主要在低温段(150~250 °C),记为γ峰,归属于化学吸附在氧空位上的O物种的还原,该氧空位可由部分Ni2+或Y3+物种扩散到CeO2晶格中形成的Ni—Ce—O或者Y—Ce—O结构产生[25]。但是除Ni/CeO2外,其余4种催化剂在此处的还原峰强度较低,可能是由于Y2O3的加入促进了Ni—C—O中的Ni被还原,从而减少了催化剂中Ni—Ce—O结构,这与XPS结果一致。250~450 °C的宽还原峰是由催化剂中NiO的还原导致的,该还原峰可根据NiO与载体相互作用强度划分为δ1和δ2,其中δ1代表与载体间相互作用较弱的NiO还原产生的还原峰[26];δ2代表与载体具相互作用较强的NiO的还原产生的还原峰[27]。从图7可知,随Y2O3含量增加,δ1和δ2还原峰面积增大且逐渐向高温迁移,表明此处的δ峰可能是由多个物种还原产生的,这可能是表面Ce物种和Ni物种还原导致[25-26],也说明Y2O3的加入改变了催化剂中活性组分Ni与Ce之间的相互作用。
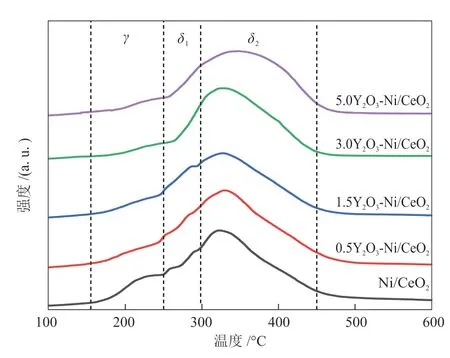
图7 xY2O3-Ni/CeO2的H2-TPR谱图Fig.7 H2-TPR spectra of xY2O3-Ni/CeO2
2.5 催化剂表面酸碱性质的分析
为了考察反应情况下催化剂表面酸碱性质,对催化剂原位还原后进行CO2-TPD测试,结果如图8所示。CO2解吸曲线可以根据温度划分50 °C<t<250 °C和250 °C<t两个区域。第1个区域内出现一个相对尖锐的脱附峰,对应催化剂表面的弱碱性位点(通常是—OH)上吸附的CO2,在低温下易于解吸脱附;第2个区域内则出现一个短而宽的脱附峰,可归属于催化剂表面的中/强碱性位点(通常是表面路易斯酸碱对)上吸附的CO2[28]。根据LIN等[29]报道,在低温下的CO2催化还原制合成天然气的反应中,归属于50~250 °C上催化剂表面弱碱性位点起到重要作用。此外可以看出在中/强碱性位点上的CO2吸附量有随Y2O3含量的增加而增加的趋势。
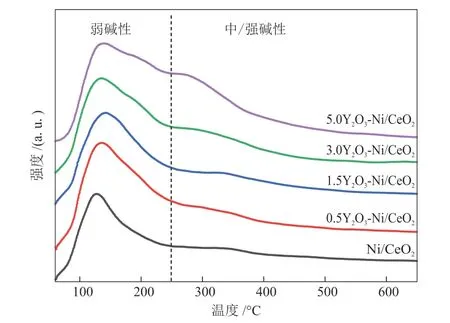
图8 还原后xY2O3-Ni/CeO2的CO2-TPD谱图Fig.8 CO2-TPD spectra of reduced xY2O3-Ni/CeO2
50~250 °C内催化剂上弱碱性位点CO2脱附峰相对面积与240 °C时催化剂上CO2转化率的关系如图9所示。可以看出,在弱碱性位点上CO2的吸附量与CO2转化率大小排列顺序保持一致。说明当加入1.5%Y2O3时,催化剂表面的弱碱性位点数量最多,同时表面的中强碱性位点数量增加,进而CO2的吸附活化得到促进,催化剂反应活性随之提升。
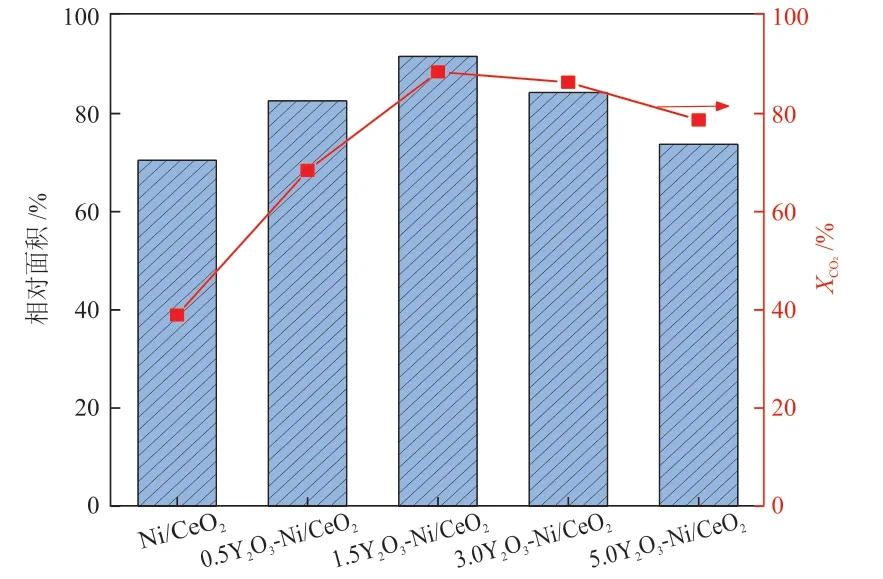
图9 催化剂上弱碱性位点CO2脱附峰相对面积与240 °C时催化剂上CO2转化率的关系Fig.9 Relationships between relative area of CO2desorption peak of weak basic sites and CO2conversion on catalysts at 240 ℃
3 结论
本文采用浸渍法和N2辉光等离子体辅助处理制备了一系列Y2O3-Ni/CeO2催化剂,考察了该处理方法下Y2O3含量对催化剂低温反应性能的影响,并对低温反应活性最优异的1.5Y2O3-Ni/CeO2进行了稳定性测试,得到以下结论。
(1)Y2O3的加入能够降低反应能垒(表观活化 能 从102.7 kJ/mol降 低 到78.8 kJ/mol),加 快反应速率,进而显著提升催化剂的低温活性。其中,1.5Y2O3-Ni/CeO2催化剂在CO2催化还原制合成天然气反应中表现出最优异的低温催化反应活性和稳定性,该催化剂在220 ℃下CO2转化率达84.1%(约为Ni/CeO2的8倍),CH4选择性达100%。
(2)Y2O3的引入减小了Ni和CeO2的粒径,增强了Ni的分散度和还原度,增加了催化剂的活性位数量,并改变了催化剂中活性组分Ni与CeO2之间的相互作用。同时调控了催化剂中氧空位含量和表面弱和中/强碱性位点含量,从而促进了CO2的低温吸附活化,提升了催化剂的低温反应活性。
猜你喜欢
分子催化(2022年1期)2022-11-02
机械工业标准化与质量(2022年6期)2022-08-12
家庭科学·新健康(2022年7期)2022-07-13
表面技术(2022年1期)2022-02-12
昆明医科大学学报(2021年3期)2021-07-22
烟草科技(2021年6期)2021-06-24
电脑知识与技术(2018年19期)2018-11-01
伴侣(2018年8期)2018-08-23
中小企业管理与科技·上旬刊(2018年12期)2018-02-18
中学化学(2015年2期)2015-06-05
