
γ-Fe2O3抗As2O3中毒能力的分子模拟
2022-08-23 09:31周文波牛胜利刘思彤韩奎华王永征
中国环境科学 2022年8期
周文波,牛胜利*,刘思彤,王 栋,韩奎华,王永征
γ-Fe2O3抗As2O3中毒能力的分子模拟
周文波1,牛胜利1*,刘思彤1,王 栋2,韩奎华1,王永征1
(1.山东大学能源与动力工程学院,高效节能及储能技术与装备山东省工程实验室,山东 济南 250061;2.佐治亚理工学院布鲁克贝尔可持续发展学院,佐治亚 亚特兰大 30332)
采用密度泛函理论研究了γ-Fe2O3表面As2O3的吸附以及掺杂改性提高抗As2O3中毒性能的作用机理.计算了As2O3在完整以及O缺陷γ-Fe2O3(001)表面的吸附性能,包括吸附位点、吸附结构、吸附能、PDOS等.同时建立了Mo、Ti、Mg掺杂的γ-Fe2O3模型,探讨了助剂掺杂对抗砷中毒能力的作用机制,并考虑了掺杂量的影响.结果表明:As2O3倾向于以O端化学吸附在γ-Fe2O3(001)表面Feoct位,吸附过程发生强烈的相互作用和电荷转移.当表面存在O缺陷时,As2O3的吸附能得到提高.Mo、Ti、Mg倾向于掺杂在Feoct位,增强了对As2O3的吸附能力,并且增大Mo的掺杂量可以强化As2O3的吸附.As2O3倾向于与活性较强的Mo、Ti、Mg发生反应,从而保护活性Fe位不受砷中毒,Ti和Mg的掺杂还抑制了相邻Fe位对As2O3的吸附.Mo、Ti、Mg的掺杂还促进了催化剂表面对NH3的吸附,增强了表面酸性强度,有利于SCR反应.Mo、Ti、Mg原子的掺杂有利于提高γ-Fe2O3催化剂的抗砷中毒性能.
As2O3;吸附;γ-Fe2O3;O缺陷;Mo、Ti、Mg掺杂;密度泛函理论
煤燃烧产生的氮氧化物会引起一系列环境问题,如酸雨和臭氧破坏等[1-3].面对巨大的环境压力和严格的排放标准,NH3选择性催化还原(NH3-SCR)仍然是主流的NO控制技术[4-5].商业化的V2O5- WO3/TiO2催化剂存在反应温度窗口窄、抗毒性差以及钒的生物毒性等诸多缺点[6-8].因此,设计更合理、更环保的中低温SCR催化剂势在必行.
由于来源广泛,环境友好,催化性能优异等优点,铁氧化物催化剂已迅速引起研究人员的关注.Liu等[9]比较了α-Fe2O3和γ-Fe2O3的催化活性和选择性,发现NO和NH3分子都更容易吸附在γ-Fe2O3表面,在150~300℃时γ-Fe2O3表现出了比α-Fe2O3更高的催化活性.张信莉等[10]制备了Mn掺杂的γ-Fe2O3催化剂,发现Mn的掺杂拓宽了脱硝活性窗口,并实现了在125~200℃效率保持100%.Xu等[11]在γ-Fe2O3催化剂中引入Mg,提高了250~350℃温度范围内的催化性能,NO转化率超过90%.上述研究充分表明,γ-Fe2O3可以成为良好的中低温SCR催化剂,但实际烟气中的一些有害成分会导致催化剂失活.
烟气中,重金属砷主要以As2O3气体形式存在,通常被认为是对SCR催化剂产生不利影响的最重要的形态[12-13].As2O3与催化剂结合阻止氨的吸附或阻塞催化剂内部通道,从而抑制NH3和NO在SCR催化剂上的活化[14].同时,As2O3能够破坏催化剂表面Lewis酸位点和Brønsted酸位点,减弱酸性位点对NH3的吸附.Li等[15]发现As2O3含量达到2.9%时,催化剂明显失活,在250~500℃时NO转化率下降到40%以下.Peng等[16]研究了As2O3对钒基催化剂失活的影响,认为主要是由于Lewis酸位点和不稳定的Brønsted酸位点数量减少所致.Li等[17]研究了砷对γ-Fe2O3催化剂的毒害行为,发现在325℃时催化剂上的NO转换率损失了44%.因此,迫切需要提高SCR催化剂的抗砷中毒能力,以提高其使用寿命和性能.
另一方面,助剂掺杂可以有效改善SCR催化剂的抗砷中毒性能.Peng等[18]比较了MoO3和WO3掺杂的V2O5/TiO2催化剂的抗砷中毒机理.研究发现,氧化砷更容易与活性较高的MoO3反应,从而保护催化剂上的活性物种.Li等[19]在γ-Fe2O3催化剂中引入Mo后发现Mo在200~400℃温度范围内提高了脱硝活性,并阻止了活性铁和气相砷的结合,对催化剂的抗砷中毒起很大的作用.通过Mg和Ti掺杂改性的γ-Fe2O3催化剂在100~350℃的催化活性和抗砷性能都明显提高[20].
由于密度泛函理论(DFT)具有在分子尺度上探测反应机理的强大功能,越来越多的研究者将其应用于催化剂砷吸附与中毒研究.Wu等[21]利用DFT探究了WO3和MoO3掺杂对钒基催化剂与As2O3相互作用的影响,发现As2O3易与MoO3发生反应,从而保护活性成分V2O5.Xing等[22]用DFT模拟发现NH3和As2O3分子在CuO(111)表面的吸附主要以物理吸附为主,As2O3比NH3更容易吸附在CuO表面.Hu等[23]利用分子模拟发现As2O3的吸附取决于所处的位置,物理吸附是由于阻碍了物理吸附向化学吸附转变的高能垒所致.然而,采用DFT研究γ-Fe2O3表面As2O3的吸附及抗中毒性能的研究很少,特别是在吸附过程中相互作用的具体微观解释还鲜有报道.
本研究采用密度泛函理论(DFT)方法研究了As2O3在γ-Fe2O3(001)表面以及缺陷表面的吸附性能和Mo、Ti、Mg掺杂对γ-Fe2O3抗砷中毒的影响.研究结果可为γ-Fe2O3催化剂的开发和耐砷中毒机理的探索提供理论参考.
1 计算模型和方法
1.1 计算模型
本研究建立了γ-Fe2O3的晶胞模型(图1).γ- Fe2O3为反尖晶石型结构,晶胞参数为===8.405Å,===90◦[24].每个晶胞含有32个O阴离子和21.33个Fe阳离子,其中八面体Fe表示为Feoct,四面体Fe表示为Fetet[25].由于γ-Fe2O3(001)面具有最强的反应活性,因此被选作反应面[26-27].如图2所示,模型选取7层原子,底部4层原子固定,顶部3层原子松弛[28].设置了P(2×2)的平面模型,以及为了防止两个周期性平板之间的相互作用,设置了15Å的真空层.
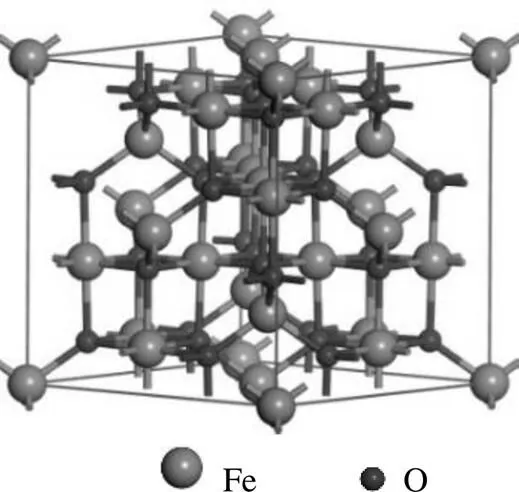
图1 γ-Fe2O3的晶胞模型
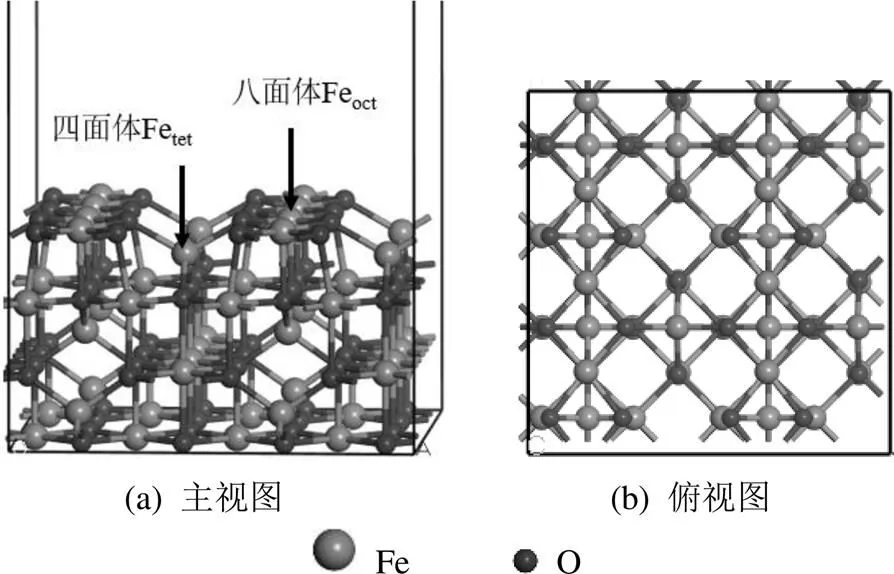
图2 γ-Fe2O3(001)表面结构

图3 几何优化的As2O3
1.2 计算方法
所有DFT计算均基于CASTEP程序包[29].扩展平面波采用具有Perdew-Burke-Ernzerhof交换相关函数的广义梯度近似(GGA-PBE)[30-31].截止能量为400eV,k点网格为2×2×1.离子核用超软赝势描述,并考虑自旋极化.结构优化收敛精度:能量为2.0×10-5eV/atom,最大应力为0.05eV/Å和最大位移为0.002Å,自洽场SCF收敛标准为2.0×10-6eV/atom.在γ-Fe2O3中,阳离子占据在四面体和八面体位置,由于四面体晶格和八面体晶格中阳离子的自旋取向为反平行排列,导致亚铁磁行为[32-33].计算允许自旋极化,以反映磁赤铁矿γ-Fe2O3的磁性,磁矩设置遵循亚铁磁性排列,四面体和八面体位点上Fe的磁矩方向相反,并采用高自旋态.
优化后γ-Fe2O3的晶格常数为8.357Å,与实验数据相符[24].在尺寸为10Å´10Å´10Å晶格中进行As2O3的几何优化,优化结果如图3所示.根据计算结果,As-O键长为1.862Å,As-O-As角为79.365°,分别与参考值1.836Å和80.6°非常接近[34].
ads定义为表面吸附能,计算见式(1):
ads=tot-ad-catal(1)
式中:ads、tot、ad和catal分别代表吸附能、气体吸附在γ-Fe2O3(001)表面的系统总能、气体分子的能量和催化剂的能量,单位eV.
Δ定义为As2O3与γ-Fe2O3(001)表面的电荷转移,计算见式(2)[35]:
Δ=ad-gas(2)
式中: Δ、ad和gas分别表示转移的电荷量、吸附态和气态小分子的电荷,单位e.
氧空位形成能计算公式如式(3)[36]:
v=OV+ 0.5O2-surf(3)
式中:v、OV、O2和surf分别代表氧空位形成能、氧缺陷表面的能量、氧分子的能量以及完美表面的总能量,单位eV.
dope定义为表面结构的掺杂能,比较不同掺杂表面的稳定性,计算见式(4)[37]:
dope=(M-Fe2O3)-(Fe2O3)+(Fe)-(M)(4)
式中:dope、(M-Fe2O3)、(Fe2O3)、(Fe)和(M)分别代表掺杂形成能、掺杂单M(Mo、Ti、Mg)原子的γ-Fe2O3表面的能量、γ-Fe2O3表面的能量、Fe和M单质晶胞内的单个原子的能量,单位eV.
2 结果与讨论
2.1 γ-Fe2O3(001)表面As2O3的吸附
2.1.1 吸附构型与吸附能 根据γ-Fe2O3(001)表面的空间位置和对称性,本文研究了As2O3分子在γ-Fe2O3(001)表面Feoct位、Fetet位、O位、hollow位和桥位5个初始吸附位点上的吸附[38].Hu等[23]通过对As2O3小分子进行分子轨道分析,发现As2O3分子的As原子端和O原子端,均可作为吸附中心.通过几何优化,根据结构特征得到了6种As2O3分子在γ-Fe2O3(001)表面的稳定构型(图4).
从表1可知,As2O3分子择优吸附的结合能大小顺序为:ads(a)>ads(c)>ads(f)>ads(d)>ads(b)>ads(e).图4中的构型(a)最稳定,As2O3分子以其O端吸附在八面体Feoct位,形成了键长为2.202Å的O-Fe键.构型(a)的吸附能为-0.75eV,电荷转移0.19e,表明其吸附过程为放热的化学吸附.此外,在构型(c)和(f)中,初始吸附位点为桥位,但优化后As2O3分子发生移动,最终分别以O端和N端吸附在Feoct位.构型(c)、(f)和(d)的吸附能分别为-0.69,-0.51和-0.50eV,属于化学吸附,其中As2O3分子的O原子端比As原子表现出更强的化学活性.在构型(b)中,As2O3分子弱化学吸附在hollow位,吸附过程为放热反应、释放能量0.41eV.在构型(e)中,As2O3分子在Fetet位的吸附,并未形成新的化学键,吸附能为-0.23eV,属于物理吸附.从吸附能分析结果表明,As2O3分子在γ-Fe2O3表面的吸附既有物理吸附又有化学吸附,与不同的吸附位点有关,Feoct原子是最佳吸附活性位.As2O3更倾向于以其O端与催化剂表面的Feoct原子进行键合.课题组前期的研究表明,γ-Fe2O3催化剂表面的八面体铁原子(Feoct)具有最好的气体吸附能力,其中NH3主要吸附到Feoct位形成吸附态NH3,其中NO的最佳吸附方式是以N端弯曲的形式吸附在Feoct顶位[39].催化剂吸附As2O3后,活性位会被部分覆盖,阻止了反应物的吸附,抑制了SCR反应.As2O3与活性铁中心结合,占据活性位点,造成失活,与Li等[17]的实验结果一致.
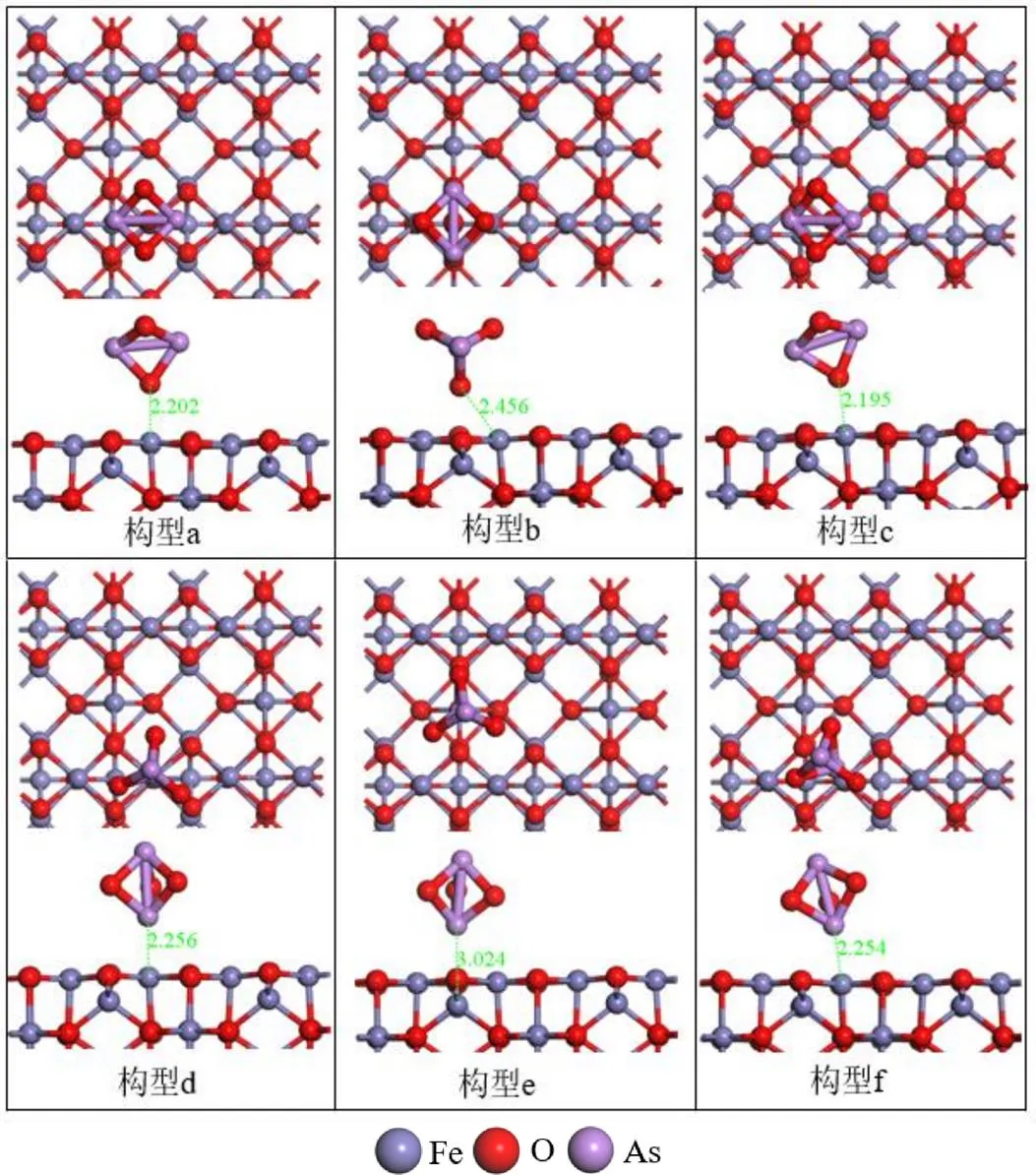
图4 As2O3在γ-Fe2O3表面的吸附构型

表1 As2O3分子吸附在γ-Fe2O3表面的优化构型
注:O-Feoct代表As2O3的O原子与Feoct原子之间的距离、As-Fe代表As2O3的As原子与Fe(Feoct或Fetet)原子之间的距离.
2.1.2 Mulliken电荷转移 通过计算Mulliken电荷布局分析,可以反映吸附过程中电荷分布与转移情况,为阐明其结构变化和吸附能提供了支持[40].从表2可知,As2O3分子在γ-Fe2O3表面的吸附过程中,除物理吸附的构型(e)外,其他构型的Mulliken电荷转移均为正数,表明吸附过程电子主要从As2O3分子转移到γ-Fe2O3表面.正如构型(a)~(c)中,当As2O3分子以O端吸附在催化剂表面时,转移的电荷主要由O原子提供,As原子的几乎没有电荷转移.当As2O3分子以As端吸附在催化剂表面时,与催化剂键合的As1原子得到电子,其他原子失去电子.As2O3分子最稳定的吸附构型(a),是以其O端吸附在Feoct原子顶位.在构型(a)的吸附过程中,电荷转移为0.19e,O1和、O2和O3分别向基底表面提供了0.05和0.08和0.05e的电子.As2O3分子中的所有O原子都失去了电子,尤其是O2原子,这是O-Fe键形成的主要因素.
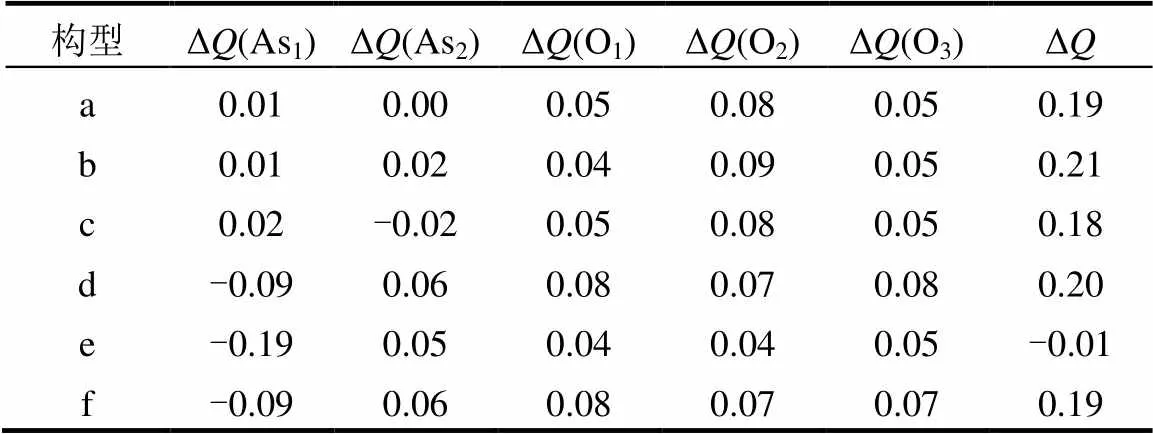
表2 As2O3吸附在催化剂表面的Mulliken电荷布局
2.1.3 偏态密度(PDOS)分析 如图5所示,在大约-5.2、-2.8、-1和2.2eV时,O原子的p轨道和Feoct的d轨道显示出杂化产生的共振峰.这表明O原子和Feoct阳离子杂化形成稳定的化学键以及稳定的吸附构型.

图5 构型a中O和Fe原子的PDOS图
O、Fe分别为As2O3分子吸附在γ-Fe2O3表面的O原子以及表面的Feoct原子,实线、划线和点划线分别为s轨道、p轨道和d轨道
综合以上分析可知,As2O3分子在γ-Fe2O3(001)表面的吸附方式,既有物理吸附也有化学吸附. As2O3分子更倾向于以其O端吸附在八面体Feoct位.As2O3分子吸附过程,会与γ-Fe2O3(001)表面发生强烈的相互作用和电荷转移.气态As2O3会与γ-Fe2O3上的活性铁中心结合,占据活性位点,导致催化剂活性下降,引起催化剂失活.
2.2 O缺陷表面As2O3的吸附
2.2.1 O缺陷的γ-Fe2O3(001)表面模型 固体材料表面通常含有空位缺陷,其中氧空位缺陷是作为催化或者化学吸附的重要活性位点,进而影响催化活性[41-42].此外,氧空位缺陷是最普遍和广泛研究的阴离子缺陷,在金属氧化物表面氧空位形成能相对较低[25].首先,在图2的γ-Fe2O3(001)表面结构的基础上,去除原结构表面Feoct与Fetet原子之间的一个氧原子,建立催化剂的氧空位模型,如图6中的O defect位置所示.氧空位的引入会使初始模型发生扭曲,导致空位附近的原子(Fe和O)出现一些显着移动.计算得出的v表明形成氧空位缺陷的可行性,γ-Fe2O3(001)(ov)的氧空位形成能v为3.56eV,表示形成氧空位缺陷时,需要外界提供能量.后续将该模型作为氧缺陷的γ-Fe2O3(001)表面结构并作为计算对象.

图6 氧缺陷的γ-Fe2O3表面结构
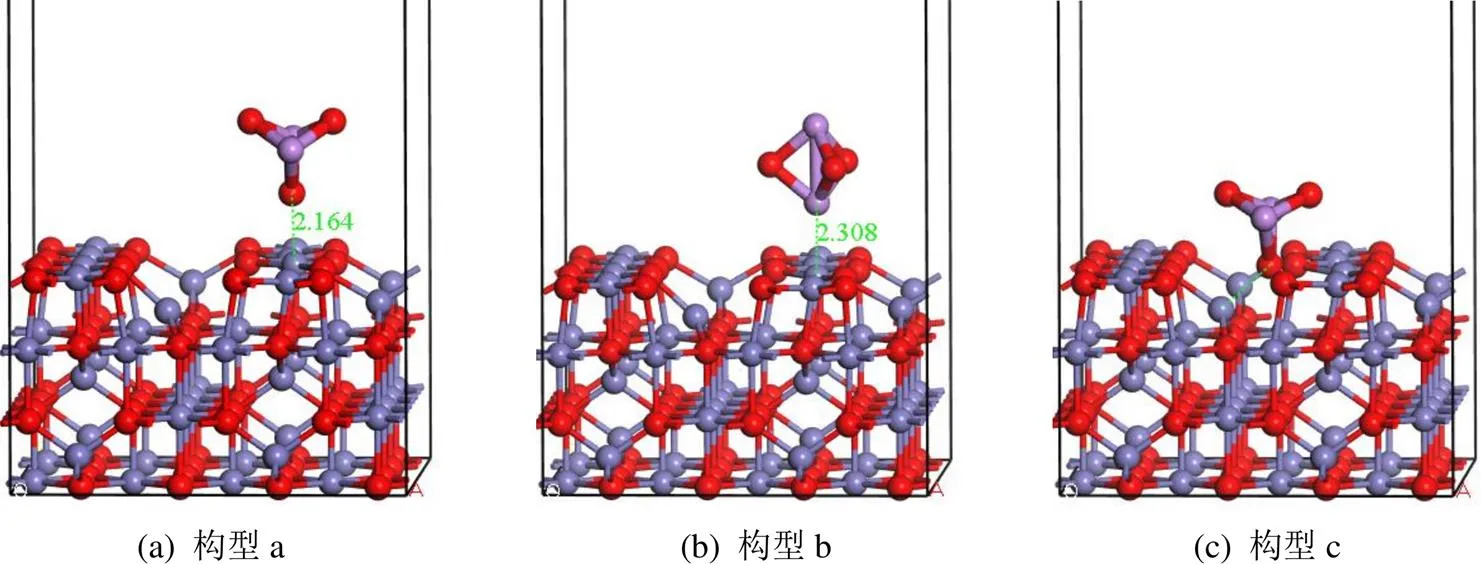
图7 As2O3在氧缺陷的γ-Fe2O3表面的吸附构型
2.2.2 As2O3在O缺陷表面的吸附 在图7中,构型(a)的结合形式几乎与图4的构型(a)相同,As2O3以O端吸附在O缺陷表面的Feoct位.构型(a)的吸附能为-0.88eV(表3),相较于在完美催化剂表面的吸附,吸附释放的能量增加了0.13eV.在这种构型下,Mulliken电荷转移为0.20e,表明在吸附过程中,一些电子从As2O3分子转移到基底.在构型(b)中,As2O3以As端吸附在O缺陷表面的Feoct位时,相较于完美表面,其吸附能的绝对值降低.在构型(c)中,As2O3以O端吸附在O缺陷的空位处,出现了新的吸附形式,O原子最终与四面体Fetet原子键合,形成了键长为1.983Å的O-Fe键.该吸附过程显著提高了催化剂表面As2O3分子的吸附能力,其吸附能-1.83eV,电荷转移为0.12e,属于化学吸附过程.综上可知,相对于完整催化剂表面,当催化剂γ-Fe2O3(001)表面存在O空位缺陷时,As2O3分子在γ-Fe2O3(001)表面的吸附能力增强.这与已有的研究结果一致:催化剂表面O空位缺陷的存在,有利于As2O3的吸附[43].

表3 As2O3在氧缺陷的γ-Fe2O3表面的优化参数
2.3 M(Mo、Ti、Mg)掺杂对γ-Fe2O3(001)表面As2O3和NH3的吸附的影响
2.3.1 M(Mo、Ti、Mg)掺杂γ-Fe2O3(001)的表面结构 已有研究表明,助剂改性可以有效改善γ-Fe2O3抗砷中毒性能[19-20].由图2可知,γ-Fe2O3(001)表面存在2种不同配位类型(八面体Feoct和四面体Fetet)的Fe原子.因此,为了构建掺杂的γ-Fe2O3(001)表面,分别使用不同的掺杂剂M(代表Mo、Ti、Mg)替换2种Fe原子(Feoct和Fetet).
如图8所示,通过结构优化得到了M-γFe2O3(001)表面模型,并计算了所有类型的M-γFe2O3(001)表面的掺杂形成能.分别对比Mo原子、Ti原子和Mg原子的不同位置的掺杂结构,发现在八面体位置的掺杂结构的比四面体位置更稳定,Mo、Ti、Mg更倾向的掺杂位置均是Feoct位.在后续研究中,将以Mo、Ti和Mg原子掺杂在Feoct位的M-γFe2O3(001)进行分析.

图8 掺杂改性后催化剂表面结构俯视图
2.3.2 M(Mo、Ti、Mg)掺杂对As2O3吸附的影响 通过Mo、Ti、Mg原子掺杂,γ-Fe2O3(001)表面增加了新的活性位点,同时会影响相邻Feoct位点对As2O3的吸附性能.基于上文的研究结果,As2O3更倾向于以其O端与γ-Fe2O3催化剂表面的Feoct原子键合.如图9a和b所示,As2O3吸附在Mo位点和相邻Feoct位点上,吸附能分别为-1.30和-0.75eV(表4).吸附过程的电荷转移分别为0.22和0.19e.通过与图4(a)结构γ-Fe2O3(001)表面的吸附比较,发现Mo的邻近Feoct位对As2O3吸附能没有变化,Mo位点对As2O3气体吸附能大大提高,说明掺杂的Mo原子更容易与As2O3气体分子结合,Mo的活跃性比Fe高.通过比较As2O3分子电荷转移的变化,可以发现Mo点上获得更多的电子,相互作用更加强烈.由于As2O3与Mo相互作用更强,As2O3更倾向于与Mo结合,从而达到保护活性组分Fe位点的作用.这与Li等[19]实验结果一致:在γ-Fe2O3中引入Mo,发现Mo提高了脱硝活性,并阻止了活性铁和砷的结合,As2O3对活性组分破坏减弱,对催化剂的抗砷中毒起很大的作用.
如图9c和d所示,As2O3吸附在Ti位点和相邻Feoct位点上,吸附能分别为-1.13和-0.71eV.在吸附过程的电荷转移量分别为0.22和0.18e.相比之下,As2O3在掺Ti的γ-Fe2O3表面的吸附能明显高于未掺Ti的表面,增加了0.38eV,并且电荷转移量更多,As2O3的活化程度增强.在Ti掺杂体系中,Ti原子的相邻Feoct位点对As2O3气体的吸附能降低.这表明掺杂的Ti不仅可以优先吸附As2O3,保护活性组分Fe位点,还抑制了活性Fe位点对As2O3的吸附.Ti原子掺杂改性可以提高γ-Fe2O3催化剂的抗砷性能.
如图9e和f所示,As2O3吸附在Mg位点和相邻Feoct位点上,吸附能分别为-1.09和-0.60eV.吸附过程的电荷转移分别为0.09和0.18e.与Ti掺杂类似,通过Mg的掺杂,As2O3不仅更倾向于吸附在掺杂的Mg位点,从而保护活性Feoct位点,还降低了As2O3在Feoct位点的吸附能.与未掺杂的γ-Fe2O3相比,Mg掺杂对相邻Feoct位As2O3的吸附能降低了0.15eV,降低效果更加明显.通过Mg掺杂可以提升γ-Fe2O3抗砷能力.
综合比较Mo、Ti、Mg对As2O3吸附的影响,发现As2O3吸附能顺序:Mo-γFe2O3(Mo)>Ti- γFe2O3(Ti)>Mg-γFe2O3(Mg)>Mo-γFe2O3(Fe)>Ti- γFe2O3(Fe)>Mg-γFe2O3(Fe),显然Mo的掺杂对As2O3吸附能力更强.综上所述,Mo、Ti、Mg原子掺杂改性均可提高对As2O3的吸附能力,保护活性Fe位点,此外通过Ti和Mg掺杂还可以抑制活性Fe位点对As2O3的吸附.Mo、Ti和Mg均是良好的抗砷中毒掺杂助剂.
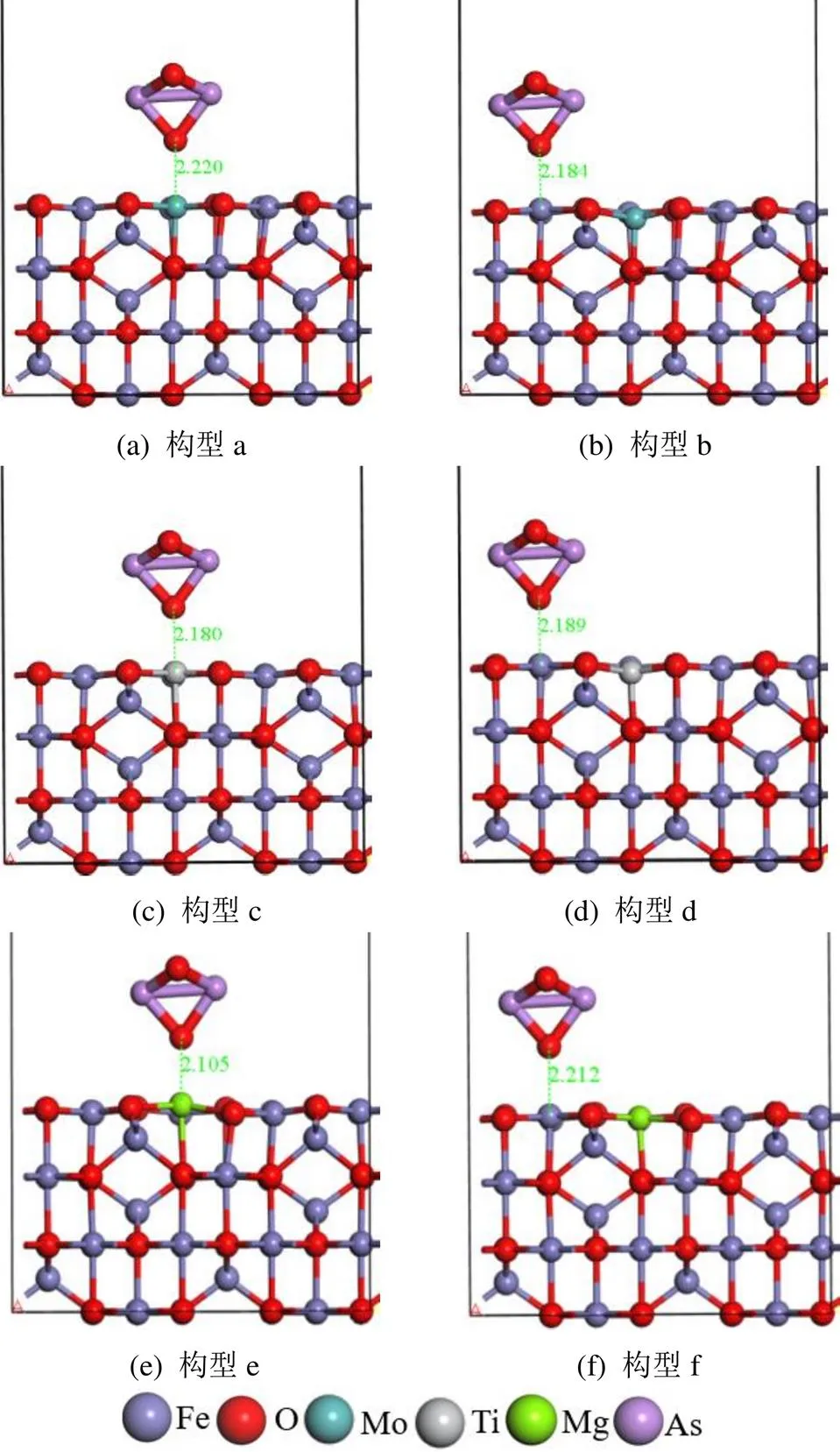
图9 As2O3在M-γFe2O3(001)表面的优化吸附模型
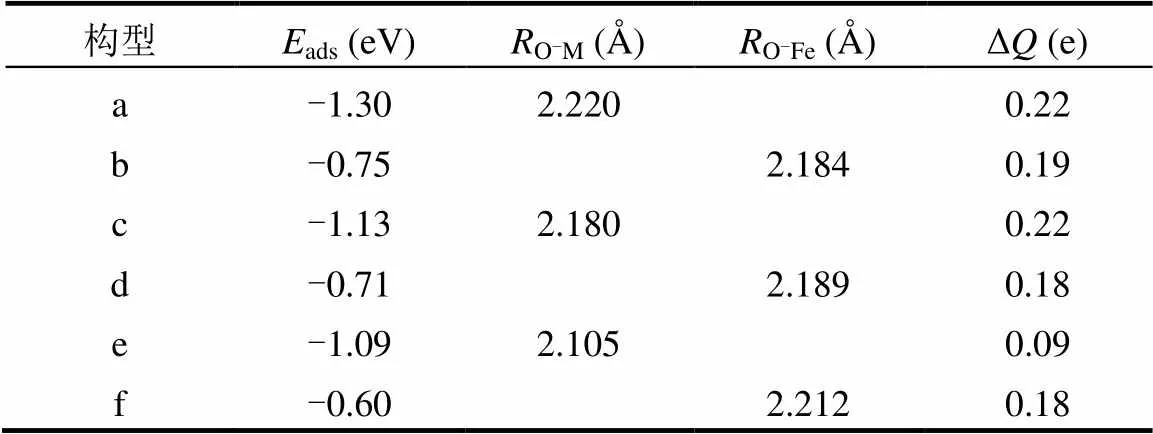
表4 As2O3在M-γFe2O3(001)表面吸附的优化参数
2.3.3 M(Mo、Ti、Mg)掺杂对NH3吸附的影响 如图10所示,首先,可以看出,NH3在γ-Fe2O3(001)表面的吸附形式是化学吸附,其吸附能为-1.01eV.还值得注意的是,在Mo、Ti、Mg原子掺杂以后,NH3在γ-Fe2O3(001)表面路易斯酸位点的吸附能值显著增加,分别增加了0.5,0.25和0.13eV.其中,通过Mo的掺杂提高催化剂酸强度最强.当NH3在Mo、Ti、Mg掺杂的Lewis酸位点上的吸附能值增加,表明NH3的吸附受到促进,吸附强度增强,这将有利于催化剂上NH3的活化.因此可以得出结论,Mo、Ti、Mg的掺杂促进了催化剂表面酸性强度的增加,有利于NH3-SCR反应.

图10 NH3在γ-Fe2O3及M掺杂表面的吸附构型

图11 As2O3在2M-γFe2O3(001)表面的吸附模型
2.3.4 掺杂量对As2O3吸附的影响 如图11a~d所示,As2O3以O端吸附在双Mo原子掺杂位置不同的2Mo-γFe2O3表面的Mo位或Fe位,掺杂位置依次是两个Mo原子分别掺杂在Fe的两个八面体位、两个四面体位以及一个四面体位和一个八面体位.对于结构(a),双Mo原子掺杂在表面双Feoct位,电荷转移到催化剂表面0.21e(表5),As2O3分子吸附能为-1.45eV,相较于单Mo原子掺杂在Feoct位的吸附能增大了0.15eV,这表明通过增大Mo的掺杂量可以强化As2O3的吸附.对于结构(c和d),双Mo原子掺杂在Feoct和Fetet位,As2O3在Mo位和相邻Feoct位,吸附能分别为-1.20和-0.75eV,电荷转移分别为0.24和0.17e,并与双Mo原子掺杂在双Feoct相比较,Mo位点吸附能明显下降.

表5 As2O3在2M-γFe2O3(001)表面吸附的优化参数
如图11e~h所示,As2O3在不同结构的2Ti-γFe2O3表面的吸附能依次为-1.14,-0.69,-1.24和-0.71eV,电荷转移分别为0.24,0.20,0.24,0.18e.对于结构(e),双Ti原子掺杂在双Feoct位,相较于未掺杂的γ-Fe2O3表面,提高了As2O3在催化剂表面的吸附能.但是,相较于单Ti原子掺杂,吸附能变化很少,增加了0.01eV.对于结构(f),双Ti原子掺杂在双Fetet位,降低了As2O3在催化剂表面活性Fe位的吸附能,有利于抗砷中毒.对于结构(g和h),双Ti原子掺杂在Feoct和Fetet位,与单Ti原子掺比,砷在Ti位点吸附能增加,Fe位点吸附能下降.Ti原子掺杂量的增加,可以更好的保护活性Fe位点,提高抗砷性能.
如图11i~l所示,As2O3在不同结构的2Mg- γFe2O3表面的吸附能依次为-0.94,-0.71,-1.08和-0.57eV,电荷转移分别为0.08,0.15,0.09,0.19e.对于结构(i),双Mg原子掺杂在催化剂表面的双Feoct位,相较于单Mg原子掺杂在Feoct位的吸附能降低了0.15eV.对于结构(k和l),双Mg原子掺杂在Feoct和Fetet位,As2O3会优先吸附在活性Mg位并在相邻Feoct位的吸附能出现明显降低,保护活性组分.
综上可知,增加Mo原子在Feoct位点的掺杂量,可以强化As2O3的吸附.通过增加Ti原子的掺杂量,可以增加As2O3在Ti位点吸附能,并降低在Fe位点吸附性能,可以更好的保护活性Fe位点,从而提高催化剂的抗As2O3性能.双Mg原子掺杂在双Feoct位,相较于单Mg原子掺杂在Feoct位的吸附能下降.
3 结论
3.1 As2O3分子在γ-Fe2O3表面的吸附既有物理吸附又有化学吸附.吸附能的不同与催化剂表面的吸附位点有关,Feoct原子是最佳吸附活性位.As2O3更倾向于以其O端吸附在γ-Fe2O3表面的Feoct位.
3.2 当γ-Fe2O3(001)表面存在O空位缺陷时,催化剂表面与As2O3分子之间存在明显的电荷转移和强相互作用,对As2O3分子的吸附能力得到了显著增强.
3.3 Mo、Ti、Mg三种改性原子倾向的掺杂位置均是γ-Fe2O3(001)表面的Feoct位.通过Mo、Ti、Mg的掺杂均可提高对As2O3的吸附能力,从而保护活性Fe位点,并且Ti和Mg的掺杂还抑制了相邻活性Fe位点对As2O3的吸附,此外增大Mo的掺杂量可以强化As2O3的吸附.通过Mo、Ti、Mg的掺杂可以增加NH3在γ-Fe2O3(001)表面的吸附,提高了催化剂表面酸强度,可以促进NH3-SCR反应.通过引入Mo、Ti、Mg对γ-Fe2O3催化剂提高抗砷中毒性能有很好的效果.
[1] 卞若愚,安忠义,李启超,等.O3-NH3协同活性焦脱硫脱硝的均相预反应特性研究 [J]. 中国环境科学,2021,41(10):4476-4483.
Bian R U,An Z Y,Li Q C,et al. Characteristics of simultaneous removal of NOand SO2by O3-NH3synergy. [J]. China Environmental Science,2021,41(10):4476-4483.
[2] Shi J,Zhang Z H,Chen M X,et al. Promotion effect of tungsten and iron co-addition on the catalytic performance of MnO/TiO2for NH3-SCR of NO[J]. Fuel,2017,210:783-789.
[3] 蒋春来,宋晓晖,钟悦之,等.2010~2015年中国燃煤电厂NO排放特征[J]. 中国环境科学,2018,38(8):2903-2910.
Jiang C L,Song X H,Zhong Y Z,et al. Characteristics of NOemissions from coal-fired power plants in China from 2010 to 2015 [J]. China Environmental Science,2018,38(8):2903-2910.
[4] Li Y H,Deng J L,Song W Y,et al. Nature of Cu species in Cu-SAPO-18catalyst for NH3-SCR: Combination of experiments and DFT calculations [J]. The Journal of Physical Chemistry C,2016,120(27):14669-14680.
[5] 付金艳,王振峰,白心蕊,等.γ-Al2O3酸性修饰稀土尾矿NH3-SCR脱硝性能[J]. 中国环境科学,2020,40(9):3741-3747.
Fu J Y,Wang Z F,Bai X R,et al. Denitration performance of NH3-SCR from γ-Al2O3acid modified rare earth tailings [J]. China Environmental Science,2020,40(9):3741-3747.
[6] 刘 晶,熊志波,周 飞,等.新型铈钨钛复合氧化物催化还原脱硝机理 [J]. 中国环境科学,2018,38(5):1670-1676.
Liu J,Xiong Z B,Zhou F,et al. The NH3-SCR mechanism of a novel cerium-tungsten-titanium mixed oxide catalyst prepared through the hydrothermal co-precipitation method modified by H2O2complex [J]. China Environmental Science,2018,38(5):1670-1676.
[7] Chen H F,Xia Y,Huang H,et al. Highly dispersed surface active species of Mn/Ce/TiW catalysts for high performance at low temperature NH3-SCR [J]. Chemical Engineering Journal,2017,330: 1195-1202.
[8] 卿梦霞,刘 亮,尹子骏,等.商用V/W/Ti系脱硝催化剂表面SO3生成的反应机理[J]. 中国环境科学,2021,41(7):3161-3168.
Qing M X,Liu L,Yin Z J,et al. Generation mechanism of SO3on the surface of commercial V/W/Ti DeNOcatalysts [J]. China Environmental Science,2021,41(7):3161-3168.
[9] Liu C X,Yang S J,Ma L,et al. Comparison on the performance of α-Fe2O3and γ-Fe2O3for selective catalytic reduction of nitrogen oxides with ammonia [J]. Catalysis Letters,2013,143(7):697-704.
[10] 张信莉,王 栋,陈莲芳,等.Mn掺杂对磁性γ-Fe2O3低温SCR脱硝活性的影响[J]. 工程热物理学报,2014,35:995-998.
Zhang X L,Wang D,Chen L F et al. Influence of Mn doping on magnetic γ-Fe2O3catalysts for selective catalytic reduction at low temperature [J]. Journal of Engineering Thermophysics,2014,35:995-998.
[11] Xu L T,Niu S L,Lu C M,et al. NH3-SCR performance and characterization over magnetic iron-magnesium mixed oxide catalysts [J]. Korean Journal of Chemical Engineering,2017,34:1576-1583.
[12] 张 月,王春波,刘慧敏,等.金属氧化物吸附剂干法脱除气相As2O3实验研究[J]. 燃料化学学报,2015,43(4):476-482.
Zhang Y,Wang C B,Liu H M et al. Removal of gas-phase As2O3in dry process by metal oxide adsorbents [J]. Journal of Fuel Chemistry and Technology,2015,43(4):476-482.
[13] 龚泓宇,胡红云,刘慧敏,等.燃煤过程中砷的迁移转化及控制技术综述[J]. 中国电机工程学报,2020,40(22):15.
Gong H Y,Hu H Y,Liu H M et al. Review of arsenic transformation and emission control during coal combustion [J]. Proceedings of the CSEE,2020,40(22):15.
[14] Ren S,Li S L,Su Z H,et al. Poisoning effects of KCl and As2O3on selective catalytic reduction of NO with NH3over Mn-Ce/AC catalysts at low temperature [J]. Chemical Engineering Journal,2018,351:540-547.
[15] Li X,Li J H,Peng Y,et al. Regeneration of commercial SCR Catalysts: Probing the existing forms of arsenic oxide [J]. Environmental Science & Technology,2015,49(16):9971-9978.
[16] Peng Y,Li J H,Si W Z,et al. Insight into deactivation of commercial SCR catalyst by arsenic: An experiment and DFT study [J]. Environmental Science & Technology,2014,48(23):13895-13900.
[17] Li X Y,Chen J,Xiao Y,et al. Insight into the homogenous and heterogeneous transformation behavior of arsenic on commercial V2O5-WO3-TiO2and novel γ-Fe2O3catalysts during selective catalytic reduction of NO[J]. Fuel,2021,301:121051.
[18] Peng Y,Si W Z,Li X,et al. Comparison of MoO3and WO3on arsenic poisoning V2O5/TiO2catalyst: DRIFTS and DFT study [J]. Applied Catalysis B: Environmental,2016,181:692-698.
[19] Li X Y,Chen J,Lu C M,et al. Performance of Mo modified γ-Fe2O3catalyst for selective catalytic reduction of NOwith ammonia: Presence of arsenic in flue gas [J]. Fuel,2021,294:120552.
[20] Li X Y,Chen J,Chen S Y,et al. Performance of Mg-Ti modified iron-based catalyst in NH3-SCR of NO at the presence of arsenic: Influence of oxygen and temperature [J]. Journal of Industrial and Engineering Chemistry,2021,101:387-396.
[21] Wu Y W,Zhou X Y,Mi T G,et al. Effect of WO3and MoO3doping on the interaction mechanism between arsenic oxide and V2O5-based SCR catalyst: A theoretical account [J]. Molecular Catalysis,2021,499:111317.
[22] Xing J Y,Wang C B,Si T,et al. Adsorption mechanism and competitive adsorption of As2O3and NH3molecules on CuO(111) surface: a DFT study [J]. Journal of Molecular Modeling,2021,27(6):178.
[23] Hu P B,Weng Q Y,Li D L,et al. Research on the removal of As2O3by γ-Al2O3adsorption based on density functional theory [J]. Chemosphere,2020,257:127243.
[24] Jørgensen J E,Mosegaard L,Thomsen L E,et al. Formation of γ-Fe2O3nanoparticles and vacancy ordering: An in situ X-ray powder diffraction study [J]. Journal of Solid State Chemistry,2007,180(1): 180-185.
[25] Jian W,Wang S P,Zhang H X,et al. Disentangling the role of oxygen vacancies on the surface of Fe3O4and γ-Fe2O3[J]. Inorganic Chemistry Frontiers,2019,6(10):2660-2666.
[26] Ren D D,Gui K T,Gu S C. Comparison of sulfur poisoning resistance of Ce/Mn doped γ-Fe2O3(0 0 1) surface in NH3-SCR reaction with DFT method [J]. Applied Surface Science,2021,561:149847.
[27] Xie C Y,Sun Y L,Zhu B Z. The promoting mechanism of doping Mn,Co,and Ce on gas adsorption property and anti-SO2oxidation over γ-Fe2O3(001) surface: A density functional theory study [J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects,2021,628:127218.
[28] Guo P,Guo X,Zheng C G. Roles of γ-Fe2O3in fly ash for mercury removal: Results of density functional theory study [J]. Applied Surface Science,2010,256(23):6991-6996.
[29] Segall M D,Lindan P J D,Probert M J,et al. First-principles simulation: ideas,illustrations and the CASTEP code [J]. Journal of Physics: Condensed Matter,2002,14(11):2717-2744.
[30] Song Z J,Wang B,Yu J,et al. Effect of Ti doping on heterogeneous oxidation of NO over Fe3O4(1 1 1) surface by H2O2: A density functional study [J]. Chemical Engineering Journal,2018,354:517-524.
[31] Zhang Y,Liu J. Density functional theory study of arsenic adsorption on the Fe2O3(001) surface [J]. Energy & Fuels,2019,33(2):1414-1421.
[32] Bentarcurt Y L,Calatayud M,Klapp J,et al. Periodic density functional theory study of maghemite (001) surface. Structure and electronic properties [J]. Surface Science,2018,677:239-253.
[33] Grau-Crespo R,Al-Baitai A Y,Saadoune I,et al. Vacancy ordering and electronic structure of γ-Fe2O3(maghemite): a theoretical investigation [J]. Journal of Physics: Condensed Matter,2010,22 (25):255401.
[34] Da Hora G C A,Longo R L,Da Silva J B P. Calculations of structures and reaction energy profiles of As2O3and As4O6species by quantum chemical methods [J]. International journal of quantum chemistry,2012,112(20):3320-3324.
[35] Lyu Z K,Niu S L,Lu C M,et al. A density functional theory study on the selective catalytic reduction of NO by NH3reactivity of α-Fe2O3(0 0 1) catalyst doped by Mn,Ti,Cr and Ni [J]. Fuel,2020,267:117147.
[36] Maitarad P,Han J,Namuangruk S,et al. Theoretical guidance and experimental confirmation on catalytic tendency of M-CeO2(M = Zr,Mn,Ru or Cu) for NH3-SCR of NO [J]. Molecular simulation,2017,43(13-16):1240-1246.
[37] 杨 涛,曹 蕃,刘利军,等.掺杂Ce/Zr对γ-Al2O3(110)表面的影响[J]. 燃烧科学与技术,2017,23(6):542-546.
Yang T,Cao F,Liu L J et al. Impact of Ce/Zr Doping on γ-Al2O3(110) surface [J]. Journal of Combustion Science and Technology,2017,23(6):542-546.
[38] Ren D D,Gui K T. Study of the adsorption of NH3and NOon the nano-γFe2O3(001) surface with density functional theory [J]. Applied Surface Science,2019,487:171-179.
[39] 周文波,牛胜利,王 栋,等.钛改性对γ-Fe2O3选择催化还原脱硝性能强化机制的分子模拟研究[J]. 燃料化学学报,2020,48(10): 1224-1235.
Zhou W B,Niu S L,Wang D,et al. Promoting effect of Ti in the Ti-modified γ-Fe2O3catalyst on its performance in the selective catalytic reduction of NO with ammonia,a DFT calculation study [J]. Journal of Fuel Chemistry and Technology,2020,48(10):1224-1235.
[40] Li Z P,Niu S L,Han K H,et al. Investigation into influences of methanol pre-adsorption on CaO(100) surface in transesterification for biodiesel production with molecular simulation [J]. Applied Catalysis A: General,2021,609:117908.
[41] Li F F,Shi C M,Wang X F,et al. The important role of oxygen defect for NO gas-sensing behavior of α-Fe2O3(0 0 1) surface: Predicted by density functional theory [J]. Computational Materials Science,2018,146:1-8.
[42] Feng Y C,Wang N N,Guo X. Density functional theory study on improved reactivity of alkali-doped Fe2O3oxygen carriers for chemical looping hydrogen production [J]. Fuel,2019,236:1057-1064.
[43] Zhang K H,Hu L T,Wang C F,et al. Middle-low-temperature oxidation and adsorption of arsenic from flue gas by Fe–Ce-based composite catalyst [J]. Chemosphere,2022,288:132425.
[44] Peng Y,Yu W W,Su W K,et al. An experimental and DFT study of the adsorption and oxidation of NH3on a CeO2catalyst modified by Fe,Mn,La and Y [J]. Catalysis Today,2015,242:300-307.
[45] Yang S J,Li J H,Wang C Z,et al. Fe-Ti spinel for the selective catalytic reduction of NO with NH3: Mechanism and structure–activity relationship [J]. Applied Catalysis B: Environmental,2012,117- 118:73-80.
[46] Man I C,Soriga S G,Parvulescu V. Effect of Ca and Sr in MgO(100) on the activation of methanol and methyl acetate [J]. Catalysis Today,2018,306:207-214.
致谢:本论文的科学计算得到了山东大学的高性能计算云平台的计算支持和帮助.
Molecular simulation study on the anti-As2O3poisoning ability of γ-Fe2O3.
ZHOU Wen-bo1,NIU Sheng-li1*,LIU Si-tong1,WANG Dong2,HAN Kui-hua1,WANG Yong-zheng1
(1.Shandong Engineering Laboratory for High-efficiency Energy Conservation and Energy Storage Technology & Equipment,School of Energy and Power Engineering,Shandong University,Jinan 250061,China;2.Brook Byers Institute for Sustainable Systems and School of Civil and Environmental Engineering,Georgia Institute of Technology,Atlanta 30332,United States).,2022,42(8):3600~3609
The γ-Fe2O3catalyst,due to its advantages in low cost and high catalytic performance,is thought to be a promising medium-low temperature SCR catalyst,but the As2O3in the flue gas likely becomes seriously deactivated. In this study,the density functional theory was used to characterize the adsorption of As2O3on the γ-Fe2O3surface as well as the mechanism of doping modification to improve the anti-As2O3poisoning performance. The adsorption properties of As2O3on the intact and O-deficient γ-Fe2O3(001) surfaces were examined,including adsorption site,adsorption structure,adsorption energy,PDOS,etc. At the same time,the catalyst model of γ-Fe2O3doped with Mo,Ti,and Mg was established to understand the mechanism of doping additives on improving the resistance to arsenic poisoning. The results show that the As2O3tends to be chemically adsorbed on Feoctsites on the γ-Fe2O3(001) surface with the O-terminus,and strong interaction and electron transfer occur during the adsorption process. When there are O defects on the surface,the adsorption energy of As2O3molecules increases. Mo,Ti,and Mg tend to be doped in Feoctsites,which thus enhances the adsorption capacity of As2O3. Increasing the doping amount of Mo can promote the adsorption of As2O3. As2O3tends to react with the more active Mo,Ti,and Mg,thereby protecting the active Fe sites from arsenic poisoning. The doping of Ti and Mg also inhibits the adsorption of As2O3on adjacent Fe sites. The doping of Mo,Ti,and Mg also promotes the adsorption of NH3on the catalyst surface and elevates the acidity of the surface,which is beneficial to the SCR reaction and to improving the anti-arsenic poisoning performance of the γ-Fe2O3catalyst.
As2O3;adsorption;γ-Fe2O3;O defect;Mo,Ti,and Mg doping;density functional theory (DFT)
X511
A
1000-6923(2022)08-3600-10
2022-01-19
山东省重大科技创新工程项目(2019JZZY020305)
* 责任作者,教授,nsl@sdu.edu.cn
周文波(1997-),男,山东青岛人,山东大学硕士研究生,主要从事燃烧与污染物控制等领域研究.发表论文2篇.
猜你喜欢
物理学报(2022年17期)2022-09-14
军民两用技术与产品(2022年1期)2022-06-01
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
作文中学版(2018年1期)2018-11-28
电子制作(2018年11期)2018-08-04
北京航空航天大学学报(2017年10期)2017-04-20
读者欣赏(2014年6期)2014-07-03
