
Sabin 株脊髓灰质炎灭活疫苗Vero 细胞残余DNA 荧光定量PCR 检测方法及其相应质量标准的建立
2022-08-17 09:39江征刘悦越朱文慧沈泓李炎郭航炜王剑锋英志芳王晓娟李长贵
中国生物制品学杂志 2022年8期
江征 ,刘悦越 ,朱文慧 ,沈泓 ,李炎 ,郭航炜 ,王剑锋 ,英志芳,王晓娟,李长贵
1.中国食品药品检定研究院,北京 102629;2.浙江省食品药品检验研究院,浙江 杭州 310052;3.四川省药品检验研究院,四川 成都 611731;4.国家药典委员会,北京 100061
脊髓灰质炎(简称脊灰)又称“小儿麻痹症”,是由3 种血清型脊灰病毒感染引起的急性传染病,主要影响5岁以下儿童,严重时造成弛缓性瘫痪。全球消除脊灰行动自1988年开展以来,通过大规模接种口服脊灰减毒活疫苗(oral live attenuated poliomyelitis vaccine,OPV)和脊灰灭活疫苗(inactivated poliomyelitis vaccine,IPV),脊灰病例数减少了 99.9%以上。2 型和3 型野生脊灰病毒相继在2015 和2019年被确认已全球消灭,目前仅剩1 型野生脊灰病毒和疫苗衍生脊灰病毒。新型冠状病毒全球大流行加剧了脊灰病毒传播,各种脊灰病例数从2018年的138 例增加至 2020年的 1 226 例[1-3]。在消灭脊灰的最后阶段,我们依然面临着挑战。
使用Sabin 减毒株生产的灭活疫苗,作为唯一一种已获批用于常规免疫的新IPV,具有避免泄露风险、全程接种不存在疫苗相关脊灰病例和疫苗衍生脊灰病毒流行、经济有效和较高可及性等优势,在全球消灭脊灰最后阶段和根除后保持较高国际市场竞争力。中国自2015年将单剂量的sIPV 引入基础免疫程序中,目前已有3 家公司获得了国家生产和销售许可证,其中2 家公司产品已通过世界卫生组织预认证;2 家公司已经进入后临床研究阶段。预计2021年以后,我国sIPV 将是世界儿童获得脊灰免疫的主力军。
sIPV 是用脊灰病毒Ⅰ、Ⅱ、Ⅲ型Sabin 株(或衍生株)分别接种于Vero 细胞,经病毒培养、收获、浓缩、纯化、灭活、按比例混合后制成。作为哺乳动物传代细胞系,Vero 细胞残留在疫苗成品中微量或少量的核酸具有潜在传染性和感染性风险,因此成品中宿主细胞核酸残留量应是关键质控项目。国际上不同监管机构基于逐个案例具体分析的风险评估方式,建立了不同疫苗产品Vero 细胞DNA 残留量限度标准,而针对sIPV 产品仍没有专门的国际标准,《中国药典》三部(2020 版)首次纳入sIPV 各论,其中规定采用通则3407 第一法(杂交法)对成品Vero细胞DNA 残留进行检测,限度应不高于50 pg / 剂,比国际上传统脊灰灭活疫苗(conventionalin activated poliomyelitis vaccine,cIPV)的限度标准(不高于100 pg / 剂)要求严格。经典的宿主细胞核酸残余检测方法包括杂交法、阈值法和荧光定量PCR(q-PCR)法,其中q-PCR 方法具有更佳的灵敏度和特异性,具有操作简便、快速、高通量等优势,成为《美国药典》(42 版)收载并在国际上普遍采用的检测手段。目前,该方法已正式收录入《中国药典》三部(2020 版)通则3407 外源性DNA 残留量测定法第三法[4-8]。本研究在《中国药典》三部(2020 版)通则3407 的基础上,建立了适用于sIPV Vero 细胞残余DNA 检测的q-PCR 方法及相应的限度标准,以弥补sIPV 成品现有传统杂交法操作繁琐、半定量等不足,达到与国际检测方法接轨的目的。
1 材料与方法
1.1 标准品、质控品及样品 Vero DNA 定量国家标准品(q-PCR 法)由中国食品药品检定研究院(简称中检院)虫媒病毒疫苗室研制并提供,批号为 20190311,含量为 107 ng /μL,100 μL /支,-70 ℃及以下避光保存,临用时进行系列稀释制备标准DNA 溶液(10 μL Vero DNA 标准品加入 525 μL 稀释液使其浓度为 2 000 pg / μL 后,10 倍系列稀释至200、20、2、0.2、0.02、0.002 和 0.000 2 pg / μL);50 pg 质控品为中检院提供的Vero 细胞核酸国家标准品(杂交法,批号为250015-201101),临用时稀释至 50 pg / 500 μL 备用;疫苗检测样品为产品上市许可企业选择自家商业化规模生产且在有效期内的30 批成品疫苗,以及尚未获批上市企业在有效期内的研究用疫苗成品批次。
1.2 主要试剂及仪器 Vero 细胞残留DNA 提取试剂盒(包含蛋白酶 K、磁珠 Bictex、LH Buffer、Wash Buffer A、Wash Buffer B、W / E Buffer)、q-PCR 试剂盒[包含 q-PCR 反应预混液(2 ×)、Mn2+、正反向引物、探针(引物及探针序列与《中国药典》三部(2020版)通则3407 第三法推荐一致)]均委托北京万泰生物药业股份有限公司制备并分装;ABI®定量PCR仪(7500 或同等及以上型号)、ABI®聚丙烯材质96孔反应板(N8010560 或同等型号)、BIOplastics®八连管及盖(B69901、B57801 或同等型号)购自美国Thermo Fisher 公司;TANBead®自动化核酸提取仪(SLA-32 或同等型号)购自台湾圆点奈米技术开发有限公司。
1.3 磁珠法提取核酸 可采用手工和仪器两种提取方式。手工提取参照《中国药典》三部(2020 版)通则3407 第三法要求进行操作。本研究推荐参与验证单位使用仪器提取方式,操作可概括为:在500 μL样品和505 μL 加标样品中分别加入20 μL 蛋白酶K,混匀,55 ℃温育 5 min。96 孔深孔板第 1 / 7 列加入 30 μL 磁珠,500 μL LH Buffer,500 μL 预处理样品或 510 μL 加标样品,第 3 / 9 列加入 500 μL Wash Buffer A,第 4 / 10 列加入 500 μL Wash Buffer B,第 6 / 12 列加入 50 μL W / E Buffer,其余为空孔。在第1、6、7、12 列对应板下方安装加热条。提取程序为:第 1 / 7 列中速混匀 5 min,磁吸 100 s;第 3 / 9列中速洗涤 4 min,磁吸 100 s;第 4 / 10 列中速洗涤3 min,磁吸 100 s,晾干 3 min;第 6 / 12 列中速洗涤2 min,将磁珠吸附100 s 后转移至3 / 9。收集第6或12 列所得溶液即为DNA 纯化液。
1.4 q-PCR 法 将q-PCR 预混液(2 ×)、Mn2(+20 ×)、引物、探针提前30 min 取出,置4 ℃融化,轻微振荡混匀,离心 3 s;分别取预混液 10 μL 及 Mn2+、引物、探针各 1 μL,配制qPCR MIX,加至PCR 八连管或96 孔板中,15 μL / 孔,再加入标准品或待测样品,5 μL / 孔,快速离心 3 s 后放入 qPCR 仪,每个样品设3个复孔,另取无核酸酶水做3个复孔,为无模板的阴性对照。设置qPCR 仪两步法反应程序:95 ℃预变性 5 min;95 ℃ 15 s,58 ℃ 30 s,40个循环;反应体积20 μL。反应结束后,将阈值(Threshold)设置为3 ~15 次循环的荧光强度均值加10 倍标准差,或采用阴性对照荧光值的最高点作为荧光阈值。读取标准曲线的斜率、截距、R2以及样品检测值。按下式计算回收率。判定试验成立条件:以至少5个连续标准品溶液浓度点生成标准曲线,R2应≥0.98,斜率应在-3.1 ~-3.8 范围内;标准品溶液浓度最低点的Ct 值不应高于39;阴性对照组如果有Ct 值时,不得低于标准品溶液浓度最低点的Ct 值;每组加标样品的回收率应在50% ~150%之间,RSD ≤ 30%。
回收率(%)=(加标样品测定值 - 样品测定值)/ 加标标准品理论值× 100%
1.5 研究设计 分为2个阶段。第一阶段,本实验室对磁珠法提取核酸以及PCR 反应体系和反应条件进行优化,对sIPV 产品进行线性范围、回收率和精密度验证;第二阶段,开展协作研究,要求5 家企业分别使用本实验室统一提供的检测试剂和q-PCR法操作步骤(简称统一方法),以及各实验室自建q-PCR 法和杂交法操作步骤对自家产品平行检测;本实验室及四川省药品检验研究院、浙江省食品药品检验研究院实验室分别使用统一方法对相应企业产品进行复核检测。要求对所有样品进行独立重复检测3 次。所有研究数据,包括每次试验是否成立的结论、50 pg 质控品检测结果及所有sIPV 产品不同方法检测结果等,汇总至本实验室,使用SPSS®Statistics 28.0 进行统一计算和分析。
2 结 果
2.1 sIPV 适用的q-PCR 法的建立及验证 采用磁珠法提取核酸,使用通则建议的Vero 细胞核酸引物和探针,优化建立了适用于sIPV 残余DNA q-PCR检测方法。反应体系(总体积20 μL):q-PCR Premix(2 ×)10 μL,Mn2(+20 ×)1 μL,正反引物(10 μmol / L)各 1.5 μL,探针(10 μmol / L)1 μL,模板 5 μL。反应条件:95 ℃预处理 5 min,1个循环;95 ℃变性 15 s,58 ℃退火30 s,共40个循环。对sIPV 产品进行方法验证,结果显示,Vero 细胞DNA 标准品在0.002 ~200 pg / μL 范围内,线性关系良好,R2= 0.999,见图1;精密度验证试验内变异系数为16.7%,试验间变异系数为27.0%,见表1;各样品回收率分别为140.8%、140.0%和117.8%,均在50% ~150%范围内,RSD 分别为11.3%、7.6%和5.5%,均 ≤ 30%,符合《中国药典》三部(2020 版)通则3407 第三法规定。
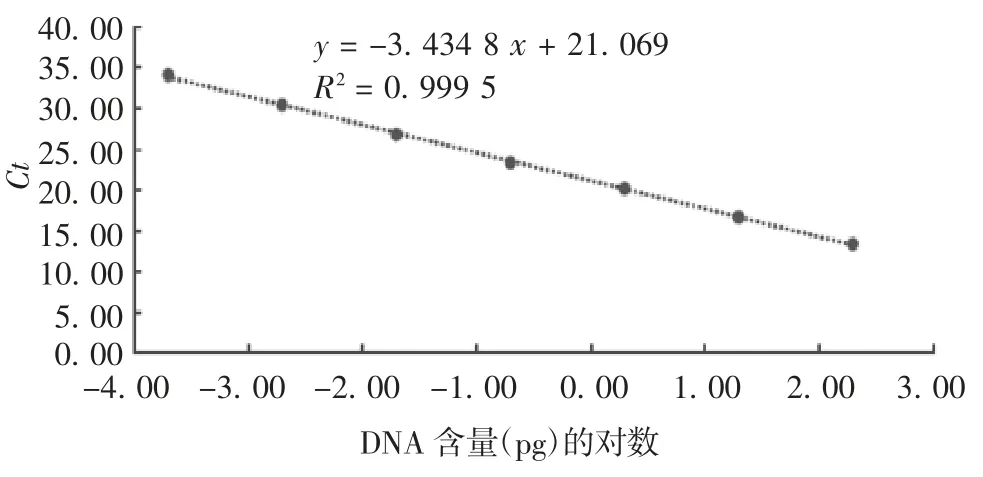
图1 sIPV 产品Vero 细胞残余DNA q-PCR 法标准曲线Fig.1 Standard curve of q-PCR method for determination of residual Vero cell DNA in sIPV

表1 sIPV 产品Vero 细胞残余DNA q-PCR 检测方法精密度验证Tab.1 Verification for precision of q-PCR method for determination of residual Vero cell DNA in sIPV
2.2 协作研究数据接收情况 所有参加协作研究单位按本研究设计要求反馈数据。本实验室汇总所有研究数据,对每一个实验室采用统一方法的试验成立情况进行逐一核对,对所有研究样品的有效检测结果进行汇总,见表2,所有实验室的试验均满足试验有效性条件,该q-PCR 方法在各实验室具有良好的适用性。
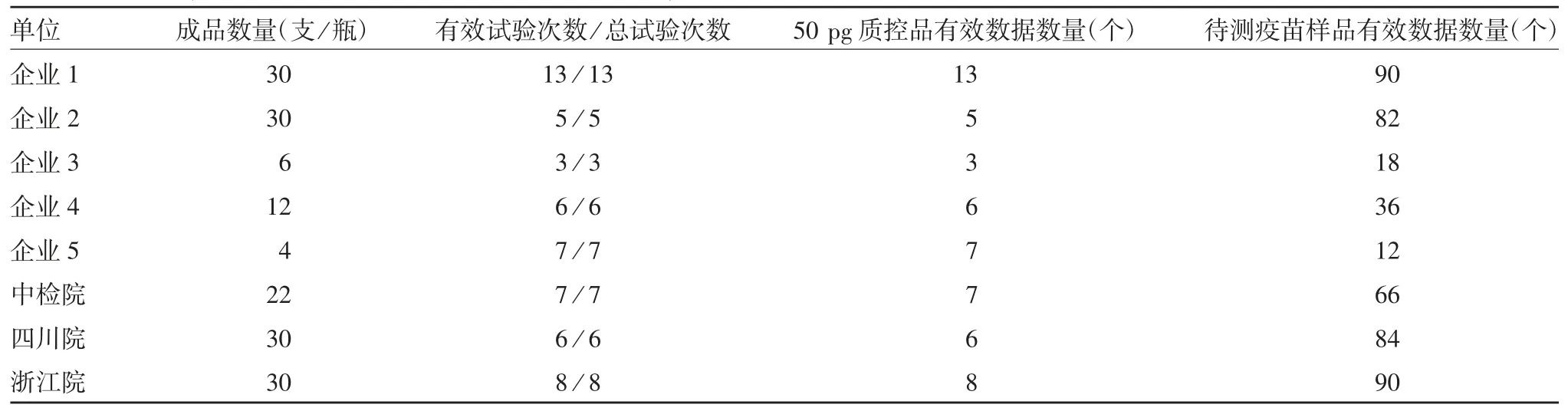
表2 协作研究接收数据汇总表Tab.2 Summary of data collected from collaborative study
2.3 50 pg 质控品协作检测统计结果 汇总所有理论值为50 pg Vero 细胞DNA 质控品的有效检测数据,共55个,使用SPSS®Statistics 28.0 软件进行统计分析,结果显示,去掉1个离群值后的54个数据呈正态分布,平均值为 50.00 pg(95% CI:44.00 ~56.01 pg),标准偏差为 22.01 pg,中位数为 47.85 pg,众数为49.20 pg;经单样本t 检验,与理论值差异无统计学意义(t = 0.001,P > 0.05)。见图 2。

图2 50 pg 质控品协作检测有效数据频率分布图Fig.2 Frequency distribution of effective data of collaborative determination of 50 pg quality control sample
2.4 各协作企业使用不同方法检测结果比较分析除了企业2 未提供自建q-PCR 法检测结果外,其余所有实验室均同时反馈了所检样品的2 种q-PCR法和杂交法检测结果,见表3。分析各企业实验室检测结果发现,使用统一方法检测结果与企业自建方法检测结果较一致,同时2 种q-PCR 法检测结果与杂交法半定量检测结果区间也较一致。

表3 各企业协作研究检测结果汇总Tab.3 Results of cooperative study by various manufacturers
2.5 各单位使用统一方法检测结果比较分析 按照要求参加协作研究的各单位均使用统一方法进行检测,其中中检院对企业3 ~5 的样品进行复核检测,浙江院对企业1 的样品进行复核检测,四川院对企业2 的样品进行复核检测。结果显示,药检实验室检测结果与企业检测结果较一致,各单位检测平均值均 < 10 pg / 剂,检测最大值均 < 50 pg / 剂,见表 4。

表4 协作研究各单位采用统一方法检测结果汇总Tab.4 Summary of collaborative determination results by q-PCR method by various participants
3 讨 论
目前经典的3 种宿主细胞核酸残余检测方法中,q-PCR 法采用特定的引物和探针,因此具有可准确定量、高特异性和灵敏度等优势,最初被《美国药典》收录,并逐渐发展成为目前全球通用的方法。《中国药典》三部(2020 版)正式收录该方法,成为继杂交法、荧光染色法之外的第三法,供不同制品验证之后采用。本研究基于《中国药典》三部(2020 版)的基本要求建立了sIPV 产品Vero 细胞DNA 残余q-PCR检测方法,一方面体现了通过有效执行和实施《中国药典》,维护其科学性和权威性,实现疫苗质量控制过程中分析检测技术的升级换代;另一方面达到与国际标准协调统一的目的,有助于进一步完善我国sIPV 生产工艺监控和产品质量评价,推动更多国产疫苗满足国际认证标准并走向世界。
本研究建立的q-PCR 法,关键在于特定引物和探针的选择,基本原则为特异序列必须是合适DNA区域内的稳定序列,且该序列的回收必须能够一致地代表所有残留DNA 的回收[8-11]。Vero 细胞作为全球病毒疫苗生产中应用最广泛、可满足大规模生产的细胞系,其核酸残留检测的特定引物序列在《中国药典》三部(2020 版)通则中已有推荐,同时q-PCR法Vero 细胞DNA 定量国家标准品已建立,因此,本研究的技术路线设定为按《中国药典》三部(2020版)q-PCR 法的规定和标识的引物及探针,采用Vero细胞DNA 定量国家标准品,针对sIPV 样品进行精密度、回收率和线性范围的方法学验证,以确定《中国药典》三部(2020 版)q-PCR 方法是否适用于sIPV。本研究结果证明,通过优化条件,q-PCR 法可适用于sIPV 产品Vero 细胞残余DNA 检测。协作研究进一步考察了该检测方法在不同企业产品和实验室间的适用性,同时,协作研究提供了更多的研究数据,为建立q-PCR 法检测sIPV 中Vero 细胞DNA 残留量的限度标准奠定了基础。
本研究参照《中国药典》三部(2020 版)通则3407-DNA 探针杂交法检测sIPV Vero 细胞残余DNA,确定的限度标准为不高于50 pg / 剂,高于国际标准(100 pg/剂)要求。为考察现有不高于50 pg/剂的标准是否适用于q-PCR 法,在此次协作研究中专门将杂交法中50 pg DNA 含量的标准品设立为q-PCR法研究的质控品之一,要求各协作单位每次试验中进行检测,最终获得55个检测值,统计分析显示这些数据呈正态分布,平均值为 50.00 pg(95%CI:44.00 ~56.01 pg),与理论值差异无统计学意义(P > 0.05)。同时各实验室方法适用性研究结果显示良好,5 家企业检测各自sIPV 成品Vero 细胞DNA 残留量平均值均 < 10 pg / 剂(0.07 ~ 7.80 pg / 剂),检测结果的最大值均<50 pg / 剂。一方面表明我国生产的sIPV 产品质量较好,宿主细胞核酸去除工艺佳;另一方面显示q-PCR 法检测宿主核酸残余设立不高于50 pg / 剂的限度标准具有可行性。
志谢感谢中国医学科学院医学生物学研究所、北京生物制品研究所有限责任公司、北京科兴生物制品有限责任公司、武汉生物制品研究所有限责任公司和北京民海生物制品有限责任公司对本研究作出重要贡献
猜你喜欢
广西植物(2022年8期)2022-09-07
大众科学(2022年7期)2022-08-19
中国慈善家(2022年3期)2022-06-14
现代苏州(2022年9期)2022-05-26
文萃报·周五版(2020年24期)2020-06-22
中学生物学(2019年7期)2019-10-17
中国药房(2018年3期)2018-10-19
分析化学(2017年12期)2017-12-25
中国质量万里行(2017年10期)2017-11-08
中学生物学(2017年7期)2017-08-23
