
Genetic dissection of the powdery mildew resistance in wheat breeding line LS5082 using BSR-Seq
2022-08-16 09:25LiruWuTongZhuHuagangXinyouCaoHaoshngLiHongxingXuMngshuJiaLipiZhangJianhngSongGharMirzaghariChngLiuPngtaoMa
The Crop Journal 2022年4期
Liru Wu, Tong Zhu, Huagang H, Xinyou Cao, Haoshng Li, Hongxing Xu, Mngshu Jia,Lipi Zhang, Jianhng Song, Ghar Mirzaghari, Chng Liu,, Pngtao Ma
a School of Life Sciences, Yantai University, Yantai 264005, Shandong, China
b School of Life Sciences, Jiangsu University, Zhenjiang 212013, Jiangsu, China
c Crop Research Institute, Shandong Academy of Agriculture Sciences, Jinan 250100, Shandong, China
d State Key Laboratory of Crop Stress Adaptation and Improvement, School of Life Sciences, Henan University, Kaifeng 475004, Henan, China
e Yantai Jien Biological Science & Technology Ltd., Yantai 265100, Shandong, China
f Department of Agronomy and Plant Breeding, Faculty of Agriculture, University of Kurdistan, P. O. Box 416, Sanandaj, Iran
Keywords:Wheat powdery mildew Bulked segregant RNA-seq (BSR-Seq)PmLS5082 Differentially expressed gene (DEG)Marker-assisted selection (MAS)
A B S T R A C T Powdery mildew of wheat is a destructive disease seriously threatening yield and quality worldwide.Comprehensive dissection of new resistance-related loci/genes is necessary to control this disease.LS5082 is a Chinese wheat breeding line with resistance to powdery mildew. Genetic analysis, using the populations of LS5082 and three susceptible parents (Shannong 29, Shimai 22 and Huixianhong),indicated that a single dominant gene, tentatively designated PmLS5082, conferred seedling resistance to different Blumeria graminis f. sp. tritici (Bgt) isolates. Bulked segregant RNA-Seq was carried out to map PmLS5082 and to profile differentially expressed genes associated with PmLS5082. PmLS5082 was mapped to a 0.7 cM genetic interval on chromosome arm 2BL, which was aligned to a 0.7 Mb physical interval of 710.3-711.0 Mb. PmLS5082 differs from the known powdery mildew (Pm) resistance genes on chromosome arm 2BL based on their origin, chromosome positions and/or resistance spectrum, suggesting PmLS5082 is most likely a new Pm gene/allele.Through clusters of orthologous groups and kyoto encyclopedia of genes and genomes analyses, differentially expressed genes (DEGs) associated with PmLS5082 were profiled. Six DEGs in the PmLS5082 interval were confirmed to be associated with PmLS5082 via qPCR analysis, using an additional set of wheat samples and time-course analysis postinoculation with Bgt isolate E09. Ten closely linked markers, including two kompetitive allele-specific PCR markers,were confirmed to be suitable for marker-assisted selection of PmLS5082 in different genetic backgrounds,thus can be used to detect PmLS5082 and pyramid it with other genes in breeding programs.
1. Introduction
Common wheat (Triticum aestivumL., 2n= 6x= 42, AABBDD) is one of the most widely cultivated cereal crops worldwide[1].Yield and quality of wheat production is seriously affected by the devastating disease, powdery mildew. The causal organism of wheat powdery mildew is the fungal pathogenBlumeria graminisf. sp.tritici(Bgt) that has complex and variable virulence structures in natural populations [2-4]. Infection by theBgtpathogen can reduce chlorophyll content, affect photosynthesis, and typically decrease wheat yield by 10%-15%and up to 62%in severe cases[5].
To control this disease, fungicides are frequently used in the field, but their long-term use has led to drug resistance due to pathogenic variation withinBgtisolates [6]. In addition, the cost of fungicides and environmental pollution caused by their use also cannot be ignored[7]. In comparison, host plant resistance is considered to be the most effective and environmentally friendly means of preventing powdery mildew epidemics [8,9]. Abundant resistance gene resources are essential for the development of resistant cultivars.So far,more than 100 formally designated powdery mildew (Pm) resistance genes have been identified at 63 loci(Pm1-Pm68,Pm8is allelic toPm17,Pm18=Pm1c,Pm22=Pm1e,Pm23=Pm4c,Pm31=Pm21) in common wheat and its diverse relatives. Moreover, more than 40 temporarily designatedPmgenes/alleles have also been reported but require further confirmation or an assurance of seed availability prior to formal designation [10,11].
There are two types of resistance patterns to powdery mildew:qualitative resistance and quantitative resistance[10-13].Qualitative resistance is common and accounts for a significant proportion of the reportedPmgenes, and these genes clearly follow Mendel’s Law of Segregation. In contrast, quantitative resistance usually involves adult-plant resistance to powdery mildew. It is conferred by polygenes whose inheritance follows a normal distribution[14,15]. Comparatively speaking, qualitative resistance often provides high level resistance to powdery mildew,but has been shown to be easily defeated after extended periods in production,whereas quantitative resistance is conferred by polygenes, and is rarely overcome [12]. Together, these two forms of resistance have provided the genetic basis of powdery mildew resistance in wheat.Due to ease of selection during breeding,the focus has mainly been on thePmgenes providing qualitative resistance, but many of thePmgenes previously conferring qualitative resistance have lost their resistance[8].Consequently,there is an urgent need to utilize more effective resistance sources to increase the genetic diversity of thePmgenes.
To date, severalPmgenes have been cloned, includingPm1,Pm2,Pm3,Pm4,Pm5,Pm8,Pm17,Pm21,Pm24,Pm41,Pm60and WTK4 [16-27], and research onPmgenes has mainly focused on the isolation ofRgenes and their use in breeding. However, these resistance genes have multiple structural types [28]. Additionally,disease resistance is a complicated process, theRgene and many associated genes jointly participate in the resistance process. The underlying molecular mechanism of disease resistance needs to be clarified to support the rational use of thePmgenes. Up to now, the mechanism for resistance to powdery mildew has been mainly investigated in grapevine [29], barley [30] andArabidopsis thaliana[31]. However, resistance to wheat powdery mildew is more complex and differs from those mentioned above. Relatively little is known regarding the molecular mechanism of powdery mildew resistance in wheat. For example, only theRgenesPm1andPm4, and the transcription factorsNAC(NAM ATAF1/2 CUC2)andMYB(V-mybavian myeloblastosis viral oncogene homolog),have been analyzed in depth [20,24,32,33].
In the identification and dissection of wheat resistance genes,bulked segregant RNA-seq (BSR-seq), which combines RNA sequencing (RNA-seq) and bulked segregant analysis (BSA), provides a highly efficient and low-cost method to rapidly map anRgene and to profile the pattern of differentially expressed genes associated with thatRgene [34,35]. BSR-seq can also alleviate the adverse effects of complex genomes, especially the allohexaploid wheat genome, and help to obtain valuable results for varying characters [36].
LS5082 is a Chinese wheat breeding line with favorable agronomic traits.In the last few years,LS5082 exhibited high resistance to powdery mildew over its entire life cycle, indicating that it is a promising donor of powdery mildew resistance gene(s)for wheat.To better understand and use the powdery mildew resistance in LS5082,we report on the identification of theRgene(s)conferring powdery mildew resistance, the analysis of resistance-related genes, and the screening of markers suitable for marker-assisted selection (MAS).
2. Materials and methods
2.1. Plant materials
The wheat breeding line LS5082 was selected by Shandong Agricultural University, Tai’an, Shandong, China. It was derived from a cross of Gao 8901 (susceptible to powdery mildew) and Xiao 13 (a wheat line from Tai’an Academy of Agricultural Sciences). LS5082 was maintained at Yantai University as a germplasm resistant to wheat powdery mildew. The wheat cultivars Shannong 29, Shimai 22 and Huixianhong, susceptible to all theBgtisolates tested, were used as susceptible parents and were crossed with LS5082 to obtain F1hybrids, F2populations and F2:3families, respectively. F1hybrids, F2populations and F2:3families were used for genetic analysis of the powdery mildew resistance in LS5082. Huixianhong was also used as the source of inoculum and susceptible control. Seven resistant donors, Coker 747 (withPm6) [37], Am9/3 (withPm33) [38], Liangxing 99 (withPm52)[39], CH7086 (withPm51) [40], WE35 (withPm64) [41], Qingxinmai(withPmQ)[42]and KN0816(withPmKN0816)[43]were used as resistant controls to differentBgtisolates, and to compare their reaction patterns with that of LS5082 through multi-race response experiments. Twenty-seven susceptible cultivars from different ecological regions of China were used to evaluate the polymorphisms of closely linked markers for MAS.
2.2. Reactions to different Bgt isolates and genetic analysis
LS5082, as well as Coker 747 (withPm6), Am9/3 (withPm33),Liangxing 99 (withPm52), CH7086 (withPm51), WE35 (withPm64), Qingxinmai (withPmQ) and KN0816 (withPmKN0816),were tested against 12Bgtisolates to evaluate their resistant spectrum. For eachBgtisolate, five seeds of each genotype were sown in 72-cell rectangular trays with each cell 3×3 cm.The susceptible control Huixianhong was sown randomly, but labelled, in each tray. Each tray was then put in an independent space with high humidity (no less than 60%) to avoid cross-infection. The growing condition was set at a daily cycle of 14 h light at 24 °C and 10 h of darkness at 18°C.At the one-leaf stage,all seedlings were inoculated with fresh conidiospores.For the first 24 h after inoculation,the seedlings were put in a dark place with high humidity(no less than 60%) at 18 °C. From the second day, the growing condition was set at a daily cycle of 14 h light at 22 °C and 10 h of darkness at 18°C,under high humidity conditions(no less than 60%).When the spores were fully developed on the first leaves of the susceptible control, which occurred at 10-14 days post-inoculation, infection types(ITs)were surveyed using the 0-4 scale described by An et al.[44],in which ITs 0,1 and 2 were regarded as resistant,and 3 and 4 as susceptible. The experiment included three independent replications.
To investigate the inheritance of the powdery mildew resistance in LS5082, E09, which is a prevalentBgtisolate in the main growing areas of winter wheat in China, was initially selected to inoculate one-leaf seedlings of LS5082, Shannong 29, Shimai 22 and Huixianhong,and their F1hybrids,F2populations and F2:3families for genetic analysis. For the resistant and susceptible parents and their F1hybrids, 10 seeds of each genotype were selected for sowing; for the F2populations, 217, 182 and 187 plants of LS5082 × Shannong 29, LS5082 × Shimai 22 and LS5082 × Huixianhong were sown, respectively; and for F2:3families,210,164 and 174 F2:3families were harvested from the corresponding F2plants of LS5082× Shannong 29,LS5082× Shimai 22 and LS5082×Huixianhong,respectively,and 20 seeds of each family were sown. If the segregation ratio against the isolate E09 was consistent with that of monogenetic inheritance, other isolates avirulent on LS5082 were used to inoculate 10 randomly selected homozygous resistant, 10 randomly selected segregating and 10 randomly selected homozygous susceptible F2:3families for genetic analysis using the same procedures as mentioned above to clarify if the same gene conferred resistance to all theBgtisolates,i.e.,to confirm that no other genes in LS5082 were conferring resistance to powdery mildew. When the phenotypic data was obtained, goodness-of-fit analysis was carried out using a chisquared (χ2) test to assess deviations of the observed phenotypic data from theoretically expected segregation ratios(SPSS 16.0 software, SPSS Inc, Chicago, IL, USA) atPless than 0.05.
2.3. BSR-Seq
Based on the phenotype against theBgtisolate E09,50 resistant plants from 50 homozygous resistant F2:3families, and 50 susceptible plants from 50 homozygous susceptible F2:3families were randomly selected for isolation of their total RNA using the Spectrum Plant Total RNA kit (Sigma-Aldrich) following the manufacturer’s protocol. Equal amounts of RNA from each of the 50 resistant plants were mixed, and similarly for the 50 susceptible plants, to construct resistant and susceptible RNA bulks, respectively.An RNA Integrity Number(RIN)≥7 was considered to meet the sequencing standard. The cDNA libraries were constructed using TruSeq Stranded mRNA LTSample Prep Kit (Illumina, San Diego, CA, USA) following the manufacturer’s protocol, and the quality of the cDNA libraries was assessed using the Agilent 2100 Bioanalyzer(Agilent Technologies,Santa Clara,CA,USA).The eligible cDNA libraries were sequenced on an Illumina HiSeq sequencing platform (Illumina HiSeq4000), according to the standard protocol by Beijing Biomarker Technologies Company Limited(Beijing, China). The sequencing indicator was set as 20 Gb clean data for each bulk.
After the cDNA libraries were sequenced,the raw data were filtered. Firstly, the reads with adapter, ribosome RNA sequences,reads with N proportion ≥10, and poor-quality reads with more than 50% bases with a Q-value ≤10 were eliminated, and highquality clean data were obtained; secondly, the STAR software(https://github.com/alexdobin/STAR) was used to assemble the clean data using the reference genome of Chinese Spring (v1.1);thirdly, single nucleotide polymorphism (SNP) calling was carried out following the reference flow chart aimed at RNA-Seq by the software GATK(v3.1-1),and the SNP index values in the two bulks were calculated using the MutMap method [45] with SNPs in the susceptible bulk as the reference; fourthly, the ΔSNP index between resistant and susceptible bulks for each SNP was calculated [46] using the following formula:ΔSNP_index = (SNP_index of resistant bulk) - (SNP_index of susceptible bulk); fifthly, euclidean distance(ED)algorithm was carried out to lock the associated interval based on the ΔSNP_index using the associated threshold value of 0.21 [47]. The interval with an associated threshold value greater than 0.21 was considered to be the candidate interval[47].
2.4. Marker development and gene mapping
After the candidate interval was confirmed, the markers linked to the knownPmgenes in the candidate region were used to screen polymorphisms for a preliminary mapping of thePmgene in LS5082. To saturate markers in the target interval, the simple sequence repeat(SSR),Indel or other sequence variations between the resistant and susceptible bulks obtained from the BSR-Seq were used to develop new SSR and Indel markers suitable for gel electrophoresis detection in BMK Cloud supported by Biomarker Technologies Company Limited(Beijing,China).PCR amplification,separation and visualization of the PCR products were as follows:1 cycle at 94°C for 3 min,followed by 35 cycles at 94°C for 30 s,50-60°C(depending on specific primers)for 40 s,72°C for 40 s,and a final extension at 72 °C for 5 min. Appropriate amounts of PCR products mixed with loading buffer (98% formamide, 0.25% bromophenol,0.25%xylene cyanol and 10 mmol L-1EDTA)were separated on 8% non-denaturing polyacrylamide gels (Acrylamide:Bisacrylamide = 25:1 or 39:1) with 1× TBE buffer (90 mmol L-1tris-borate, 2 mmol L-1EDTA, pH 8.3), and visualized by silver straining as described by Ma et al. [8].
Kompetitive allele-specific PCR(KASP)markers were also developed forPmgene(s)mapping in LS5082.Firstly,distinctive SNPs in the candidate interval was screened;secondly,sequences of 100 bp upstream and downstream of the distinctive SNP were acquired for KASP marker development using both the Polymarker website(http://www.polymarker.info/) and Primer 5 software; thirdly,the amplification sequences of the designed primers were aligned once again in theTriticeaeMulti-omics Center(http://202.194.139.32/) to ensure specificity of the sequences. The KASP assays were performed on a Bio-Rad CFX real-time PCR system (Bio-Rad Laboratories, Inc, CA, USA) with a final volume of 10 μL containing 3.0 μL of genomic DNA (125 ng), 5.6 μL of 2× KASP Master Mix(provided by LGC), 0.17 μL of primer mix (balanced mix of three pairs of primers for each marker)and 1.23 μL ddH2O. The amplification procedure was set as follows:94°C for 15 min,followed by 10 touchdown cycles of 94 °C for 20 s, 64 °C to 58 °C (decreasing 0.6 °C per cycle), and 38 cycles of regular amplification (94 °C for 20 s, 58 °C for 60 s), and the final fluorescence was detected at 20 °C using Bio-Rad CFX Manager 3.1 software (Bio-Rad Laboratories, Hercules, CA, USA).
All the newly developed markers were tested for polymorphisms between resistant and susceptible parents and their derived resistant and susceptible bulks. The polymorphic markers were then genotyped on the F2:3population of LS5082 × Shannong 29. After obtaining the genotyping data, χ2test was carried out to assess the observed phenotypic data of the F2:3families from theoretically expected segregation ratios for goodness-of-fit analysis. The linkage map was constructed based on the Kosambi function in the MAPMAKER 3.0 [48,49].
2.5.Separation with the known Pm genes on the chromosome arm 2BL
To distinguish thePmgene (s) in LS5082 and the knownPmgenes on chromosome arm 2BL, 52 markers closely linked to the knownPmgenes on chromosome arm 2BL, including 37 SSR, six expressed sequence tag (EST), five sequence-tagged site (STS),and four intron targeting(IT)markers,were assessed for polymorphisms between resistant and susceptible parents and bulks derived from the F2:3families of LS5082× Shannong 29.Polymorphic markers will be used to assess the genetic diversity between thePmgene interval in LS5082 and the documentedPmgene intervals, which could also be a supplementary means of clarifying the relationship between the unknownPmgene (s) in LS5082 and the knownPmgenes on the same chromosome arm.
Allelic tests between LS5082 and each of Liangxing 99 and KN0816,which havePm52andPmKN0816,respectively,were performed using their F2populations.Bgtisolate E09,which was avirulent on LS5082, Liangxing 99 and KN0816, was used to inoculate the F2populations of LS5082×Liangxing 99 and LS5082×KN0816 using Huixianhong as the susceptible control.The number of resistant and susceptible plants was counted after phenotyping. The allelic relationships between thePmgene in LS5082 andPm52andPmKN0816were then analyzed based on the ratio of resistant and susceptible plants.
2.6. Differentially expressed gene (DEG) analysis
After aligning the clean reads (obtained from BSR-Seq) to the Chinese Spring reference genome sequenced by IWGSC (the International Wheat Genome Sequencing Consortium) (v1.1), FPKM(Fragments per kilobase of exon per million fragments mapped)was used to calculate the expression level of the reads [50]. The software EBSeq was used to detect DEGs using Fold change ≥2 and FDR (False discovery rate) less than 0.01 as thresholds.Statistical significance of DEGs was determined using a combination of multiple tests and FDR[51].Statistics and clustering analysis of DEGs between resistant and susceptible bulks were carried out to present the expression patterns on a genome-wide scale.
Functional annotation of the DEGs was performed using IWGSC database (v1.1). Clusters of orthologous groups (COG) and kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analyses were performed using anRpackage for DEGs [52]. For COG analysis,the unigene sequences were aligned to the COG database to predict possible functions,and to determine the gene function distribution characteristics (http://www.ncbi.nlm.nih.gov/COG). For KEGG pathway analysis, the KEGG database was used to blast against the metabolic pathway (https://www.kegg.jp/kegg/pathway.html).
2.7. qPCR
After analysis of the DEGs using COG and KEGG, the DEGs related to disease resistance and/or stress tolerance were selected to profile their expression patterns via qPCR. To detect transcription levels, primers for the specific DEGs were designed based on the coding sequences of the selected genes. The seedlings of LS5082 and susceptible Shimai 22 were inoculated with theBgtisolate E09 at the one-leaf stage, and the first leaves of LS5082 and Shimai 22 seedlings were sampled 3, 6, 12, 24, 36, 48 and 72 h after inoculation, with three parallel experiments. Total RNA was extracted using the Spectrum Plant Total RNA kit (Sigma-Aldrich)following the manufacturer’s recommendations,and then quantified by measuring absorbance at the wavelengths of 260 and 280 nm using a Nano Drop 1000 spectrophotometer (Thermo Scientific). Promega DNase I-treated RNA was used for cDNA synthesis using Invitrogen SuperScript-II reverse transcriptase following the manufacturer’s guidelines.The cDNA was analyzed using qPCR,following the procedure described by He et al. [3,18] using SYBR green master mix(Applied Biosystems)with a Rotor-Gene-Q(Qiagen). Amplification was followed by melt curve analysis. Relative quantification was carried out using the 2-ΔΔCTmethod [18].Oligonucleotides amplifyingACTINwere used for normalization.
2.8. Evaluation of the closely linked markers for MAS
To select the markers suitable for MAS,the closely linked markers were genotyped on LS5082 and 27 susceptible wheat cultivars from different regions of China. Polymorphisms between LS5082 and 27 susceptible wheat cultivars were analyzed,and the markers that consistently amplified polymorphic band(s) between LS5082 and these susceptible cultivars were regarded as suitable for MAS in these genetic backgrounds.To transfer thePmgene to applicable backgrounds, these cultivars were crossed with LS5082 to construct F2and F3segregation populations for MAS.
3. Results
3.1. Inheritance of the powdery mildew resistance in LS5082
After inoculated with theBgtisolate E09,LS5082 showed no visible symptoms or hypersensitivity on the first leaf, and hence it was regarded as immune (IT 0) (Table S1). On the other hand,the susceptible parents,Shannong 29,Shimai 22 and Huixianhong,showed abundant sporulation with more than 80% of the first leaf covered with aerial hyphae, and hence they were regarded as highly susceptible (IT 4). The F1plants of LS5082 × Shannong 29,LS5082 × Shimai 22 and LS5082 × Huixianhong showed similar reaction pattern as the resistant parent LS5082 with an IT 0, suggesting that thePmgene (s) in LS5082 displayed dominant inheritance. The phenotypes of all the three F2populations fitted the expected ratios of 3:1(resistant:susceptible individuals)for monogenic segregation (Table S1), suggesting that a single dominant gene is involved in the powdery mildew resistance of LS5082. To further confirm this result and validate the genotypes of the resistant F2plants,all the tested F2plants were transplanted in the field to provide F2:3families. All the F2:3families derived from LS5082 and the three susceptible parents fitted the expected ratios of 1:2:1(Homozygous resistant:segregating:homozygous susceptible families)(Table S1).Therefore,we concluded that the resistance toBgtisolate E09 in LS5082 was conferred by a single dominant gene,tentatively designatedPmLS5082.
To further confirm ifPmLS5082also confers resistance to otherBgtisolates, 10 homozygous resistant, 10 segregating, and 10 homozygous susceptible F2:3families of LS5082 × Shannong 29 were randomly selected and inoculated with eight additionalBgtisolates shown to be avirulent on LS5082.The phenotypes of these selected families were all consistent with that ofBgtisolate E09,suggesting thatPmLS5082conferred the resistance to all the testedBgtisolates.
3.2. SNP calling and confirmation of candidate interval
After BSR-Seq,37.5 and 20.2 Gb clean data were obtained from resistant and susceptible bulks, respectively. The percentages of clean reads with a Q30 were 92.68% and 92.46%, the GC contents were 56.50% and 54.68%, the percentages of reads mapping to the reference genome were 87.24%and 73.78%,respectively.These data indicated that the sequencing quality was high and suitable for subsequent analysis. A total of 47,155 high quality SNPs between resistant and susceptible bulks were detected from the clean data for subsequent ΔSNP index analysis.Using the ED algorithm and 0.21 as the threshold,only one putative candidate region near the end of chromosome arm 2BL(652.4-714.4 Mb)was identified (Fig. 1). There are 1028 SNPs in this candidate region, which were used for subsequent marker development and DEG analysis(Table S2).
3.3. Molecular mapping of PmLS5082
After the candidate region was confirmed,the markers linked toPm52,Pm63,Pm64andPmQwere used to genotype the F2:3families of LS5082 and Shannong 29. The SSR markersWGGBH612-5,WGGBH913andXicsq405were linked toPmLS5082, andPmLS5082was flanked by the markersWGGBH612-5andWGGBH913with genetic distances of 0.3 and 7.1 cM,respectively(Fig.2).The corresponding candidate region using the IWGSC Chinese Spring reference genome v1.1 was narrowed to 710.3-715.0 Mb on chromosome arm 2BL.
To saturate the linkage map and further narrow down the candidate region, new gel-based and gel-free KASP markers were designed using the BSR-Seq derived Indels, SSR, differential sequences and SNPs between resistant and susceptible bulks in the candidate region(710.3-715.0 Mb).Seven gel-based SSR markers (YTU19-005,YTU19-007,YTU19-009,YTU19-011,YTU19-012,YTU19-014andYTU19-016)(Table S3)and two gel-free KASP markersYTU19-KASP26andYTU19-KASP96showed consistent polymorphism between parents and bulks (Table S4). These markers were found to be linked withPmLS5082by genotyping the F2:3families of LS5082 and Shannong 29 (Fig. 3). All the linked markers fitted Mendel’s Law of Segregation for a single dominant gene. A highdensity linkage map was then constructed using all the linked markers above (Fig. 2).PmLS5082was flanked byWGGBH612-5andYTU19-005with genetic distances of 0.3 and 0.4 cM, respectively. The corresponding physical interval was narrowed to 710.3-711.0 Mb on the chromosome arm 2BL (Fig. 2).
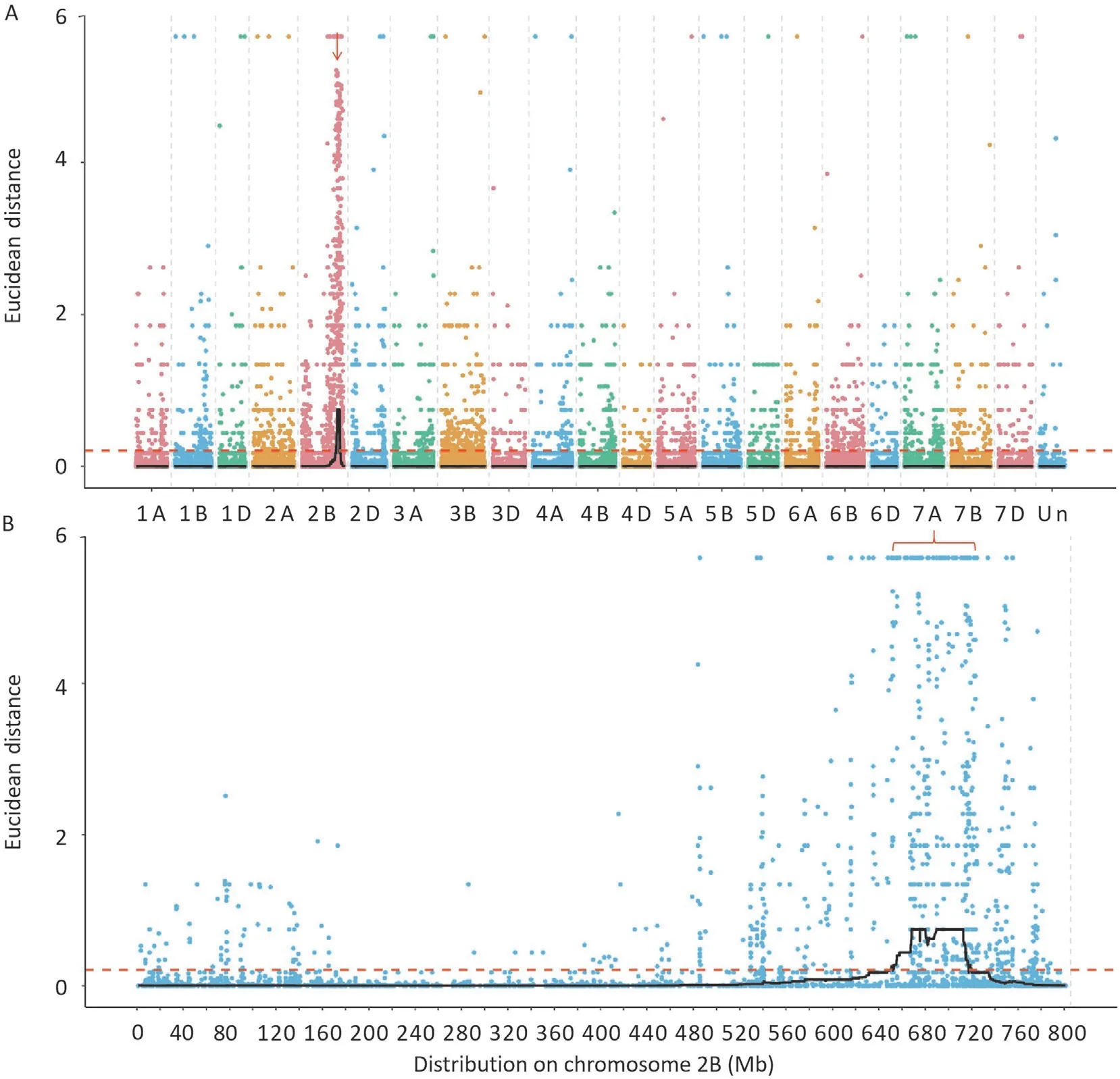
Fig.1. Distribution of the single nucleotide polymorphisms(SNPs)with differences between resistant and susceptible bulks on 21 chromosomes(A)and on chromosome arm 2B (B) based on ΔSNP index value.
3.4. Comparisons of PmLS5082 and the known Pm genes on chromosome arm 2BL
After tested against 12Bgtisolates at the seedling stage,LS5082 was resistant to 10 out of 12 (83%) of the isolates with an IT 0.LS5082 even showed an immune reaction pattern towards the highly virulent isolates E05, E20 and E21 (Fig. 4; Table S5). Meanwhile,PmLS5082showed a significantly different response spectrum to those of the knownPm6,Pm33,Pm51,Pm52,Pm64,PmQ,andPmKN0816on chromosome arm 2BL (Fig. 4; Table S3). This result suggests thatPmLS5082is most likely a newPmgene/allele.
Because LS5082, Liangxing 99 (Pm52donor) and KN0816(PmKN0816donor) are all Chinese breeding lines, allelism tests betweenPmLS5082and the reportedPm52andPmKN0816were carried out to further confirm their genetic relationship. Nine and four susceptible plants were found from 3002 and 5667 F2plants of LS5082×Liangxing 99 and LS5082×KN0816,respectively,suggesting thatPmLS5082was closely linked withPm52andPmKN0816, but different from them.
Combining the analyses of the resistant spectrum and allelism tests,PmLS5082is most likely different from the knownPmgenes on chromosome arm 2BL.
3.5. Discovery and analysis of DEGs
After calculating the expression level of the reads and following functional annotation, a total of 10,646 DEGs were identified between resistant and susceptible bulks,of which 5280 DEGs were down-regulated and 5366 DEGs were up-regulated using the expression index of susceptible bulk as a standard (Table S6;Fig. S1). COG analysis was then performed on the DEGs between resistant and susceptible bulks. The data show that the functions of the DEGs mainly involve two categories: signal transduction mechanisms, and carbohydrate transport and metabolism. The DEGs related to defense mechanisms also accounted for a noticeable proportion (Fig. S2).
To investigate further the signal transduction pathways that the DEGs may be involved in, significance enrichment analysis for KEGG pathway was performed on the DEGs between resistant and susceptible bulks. A total of 126 significantly enriched(Q ≤0.05) pathways involving 20 categories were found (Fig. S3;Table S7). Among these, one plant-pathogen interaction pathway was enriched, and 18 DEGs were present in this pathway(Fig. S4). These genes were used as targets for further molecular studies into the plant response to powdery mildew.
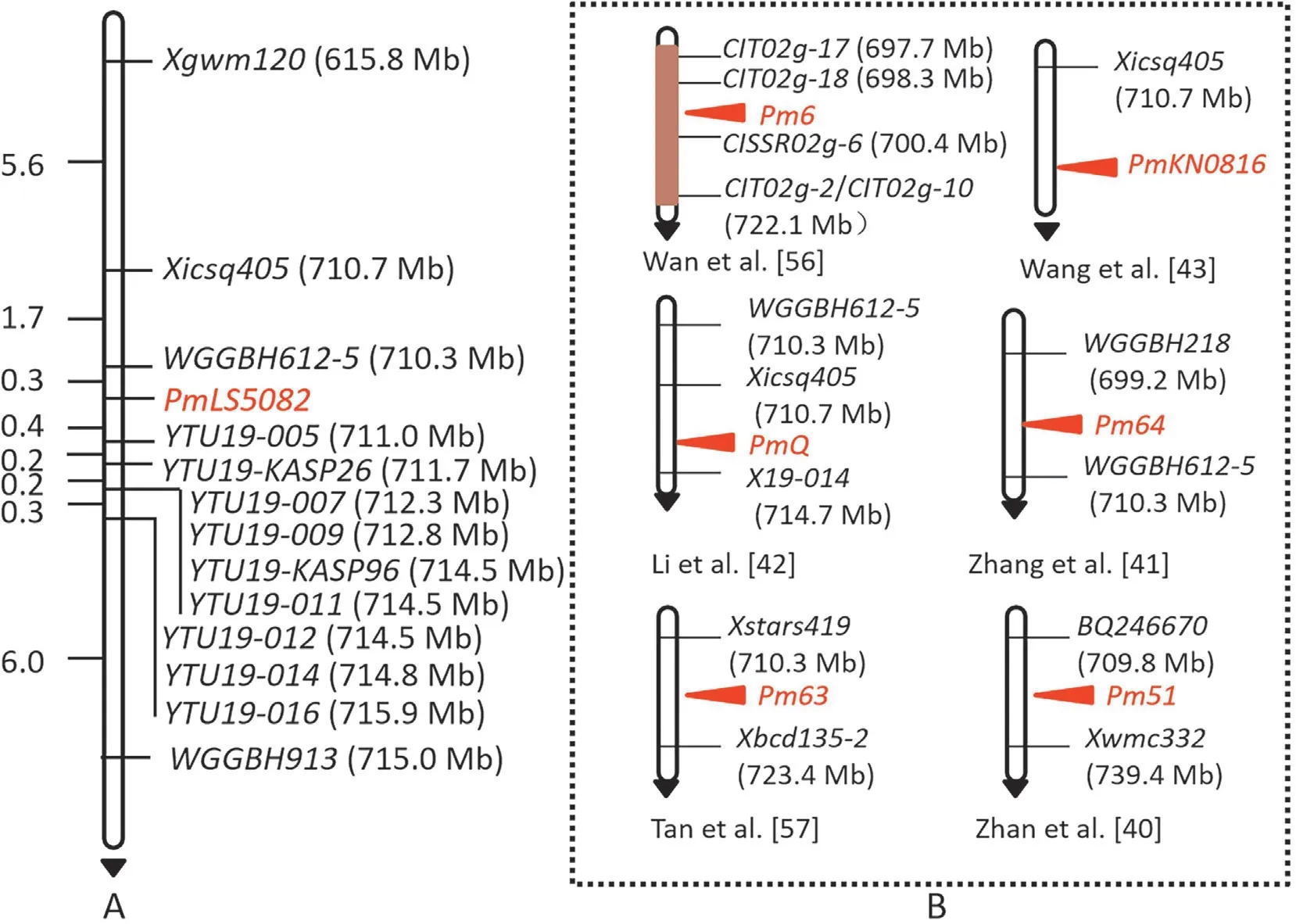
Fig. 2. Linkage map of PmLS5082 using the F2:3 families of LS5082 × Shannong 29 (A) and the physical locations of known Pm genes on chromosome arm 2BL (B). Genetic distances in cM are showed to the left. The black arrows point to the centromere.
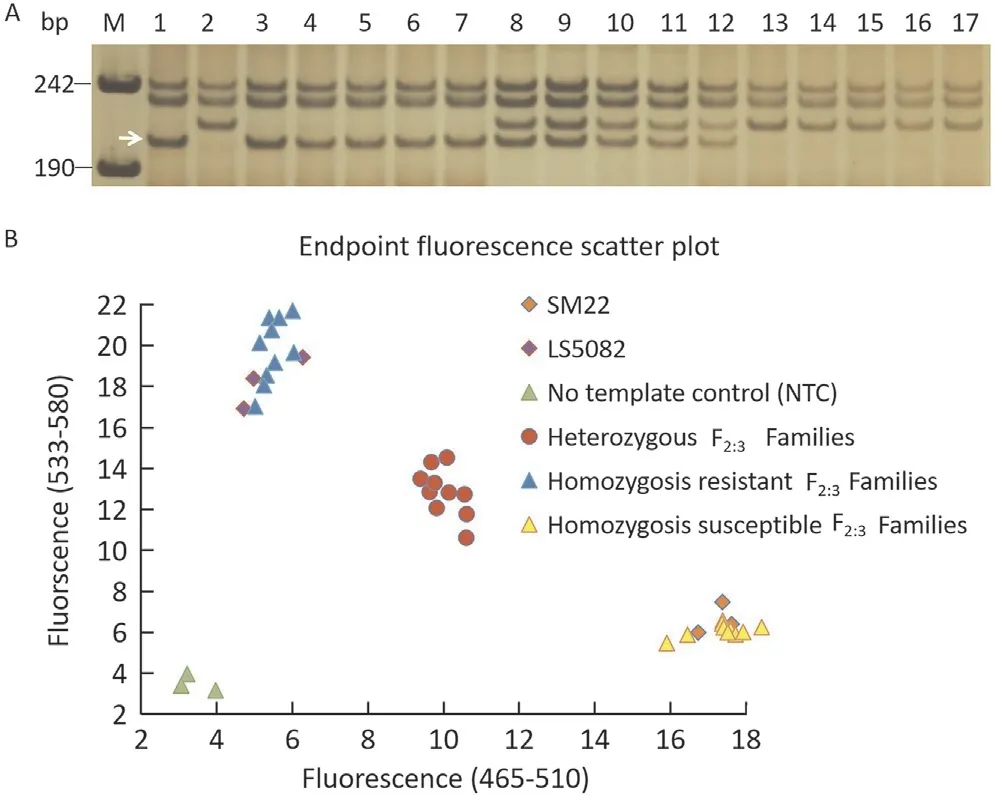
Fig. 3. Amplification patterns of PmLS5082-linked markers YTU19-005 (A) and genotyping result of the PmLS5082-linked kompetitive allele-specific PCR (KASP)marker YTU19-KASP96 (B) for LS5082, Shannong 29 and randomly selected F2:3 families of LS5082×Shannong 29.Lane M,pUC18 Msp I;Lanes 1-2,parents LS5082 and Shannong 29; Lanes 3-7, homozygous resistant F2:3 families; Lanes 8-12,homozygous susceptible F2:3 families;Lanes 13-17,heterozygous F2:3 families.The white arrows indicate the polymorphic bands in LS5082.
3.6. Expression patterns of the resistance-related genes in the candidate interval
To profile the expression of the resistance-related genes in LS5082, 13 DEGs related to disease resistance and/or stress tolerance in the candidate interval (Table S8) were selected and their transcription was monitored at different stages after inoculation withBgtisolate E09. Among these genes, six showed significant differences between LS5082 and Shimai 22 in the time course analysis followingBgtinoculation (Fig. 5). The transcript level of TraesCS2B02G512300.1 (encoding a G-type lectin S-receptor-like serine-protein) in LS5082 was rapidly up-regulated at 3-6 h and reached a peak at 24 h after inoculation.Only after 48 post inoculation did this gene start to up-regulate in Shimai 22.The transcript level of TraesCS2B02G512400.1 (encoding a receptor-like serineprotein) in LS5082 was rapidly up-regulated at 24 h after inoculation, but not in Shimai 22. The transcript level of TraesCS2B02G524300.1 (encoding an LRR receptor-like serineprotein) in LS5082 was rapidly up-regulated at 3-6 h and also 48 h after inoculation but showed low expression in Shimai 22.The transcript level of TraesCS2B02G520300.1(encoding a zinc finger CCCH domain-containing protein) in LS5082 was first upregulated at 24 h and again up-regulated at 48 h and 72 h after inoculation, but there was no corresponding up-regulation in Shimai 22. The transcript level of TraesCS2B02G480000.1 (encoding a peroxisomal biogenesis factor 6) in LS5082 was rapidly upregulated at 6 h, but only showed low expression in Shannong 29. The transcript level of TraesCS2B02G521800.1 (encoding an abscisic acid-inducible protein) in LS5082 began to up-regulate from 3 to 6 h and again up-regulated between 36 h and 48 h after inoculation, but there was no corresponding high expression in Shimai 22.Combining the expression patterns againstBgtinvasion and functional annotation, the TraesCS2B02G524300.1, that encoded an LRR receptor-like serine-protein, could be considered as the candidate gene ofPmLS5082, while the others may be regulatory genes involved in the resistance process.
3.7. Molecular markers for MAS

Fig.4. Reaction patterns of LS5082 and genotypes with known Pm genes on chromosome arm 2BL to Bgt isolates A3,E09,E18,and E21.1,susceptible cultivar Huixianhong;2,LS5082; 3, Qingxinmai (with PmQ); 4, WE35 (with Pm64); 5, CH7086 (with Pm51); 6, Am9/3 (with Pm33); 7, Liangxing 99 (with Pm52); 8, Coker 747 (Pm6); 9, KN0816(PmKN0816).
Ten gel-based (Xicsq405,WGGBH913,WGGBH612-5,YTU19-005,YTU19-007,YTU19-009,YTU19-011,YTU19-012,YTU19-014andYTU19-016) and two high-throughput KASP markers (YTU19-KASP26andYTU19-KASP96) were tested for their availability for MAS (Fig. 6; Table S9). All the markers, exceptYTU19-009andYTU19-KASP96, could detect polymorphic genotypes between LS5082 and most of the tested cultivars, suggesting that these markers can be used to for MAS ofPmLS5082when it is transferred into these susceptible cultivars by crossing. Among them, the gelbased markerYTU19-005and the high-throughput KASP markerYTU19-KASP96were particularly good at distinguishing between genotypes and could also distinguish homozygous and heterozygous genotypes, making them highly suitable for MAS (Fig. 6). To further verify the usefulness of these two markers, we also used them to discriminate between the breeding populations of LS5082 and five susceptible cultivars Yannong 1212, Tainong 18,Liangxing 619, Jimai 229 and Shimai 15. Among a set of 50 F2plants from LS5082 × Yannong 1212, LS5082 × Tainong 18 and LS5082 × Liangxing 619, and a set of 50 F3plants from each of LS5082 × Jimai 229 and LS5082 × Shimai 15, only one plant from each of LS5082×Tainong 18 and one plant from LS5082×Shimai 15 showed inconsistencies genotype and phenotype. These two plants were most likely recombinants. In conclusion, the gelbased markerYTU19-005and the high-throughput KASP markerYTU19-KASP96were suitable for MAS ofPmLS5082and provided different types of markers to meet the requirements of different detection platforms.
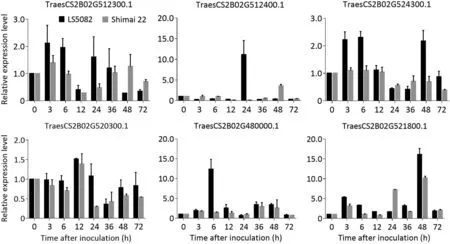
Fig. 5. Expression profiles of TraesCS2B02G512300.1, TraesCS2B02G512400.1, TraesCS2B02G524300.1, TraesCS2B02G520300.1, TraesCS2B02G480000.1, and TraesCS2B02G521800.1 in each corresponding stage of LS5082 and Shannong 29 after Blumeria graminis f. sp. tritici (Bgt) infection.
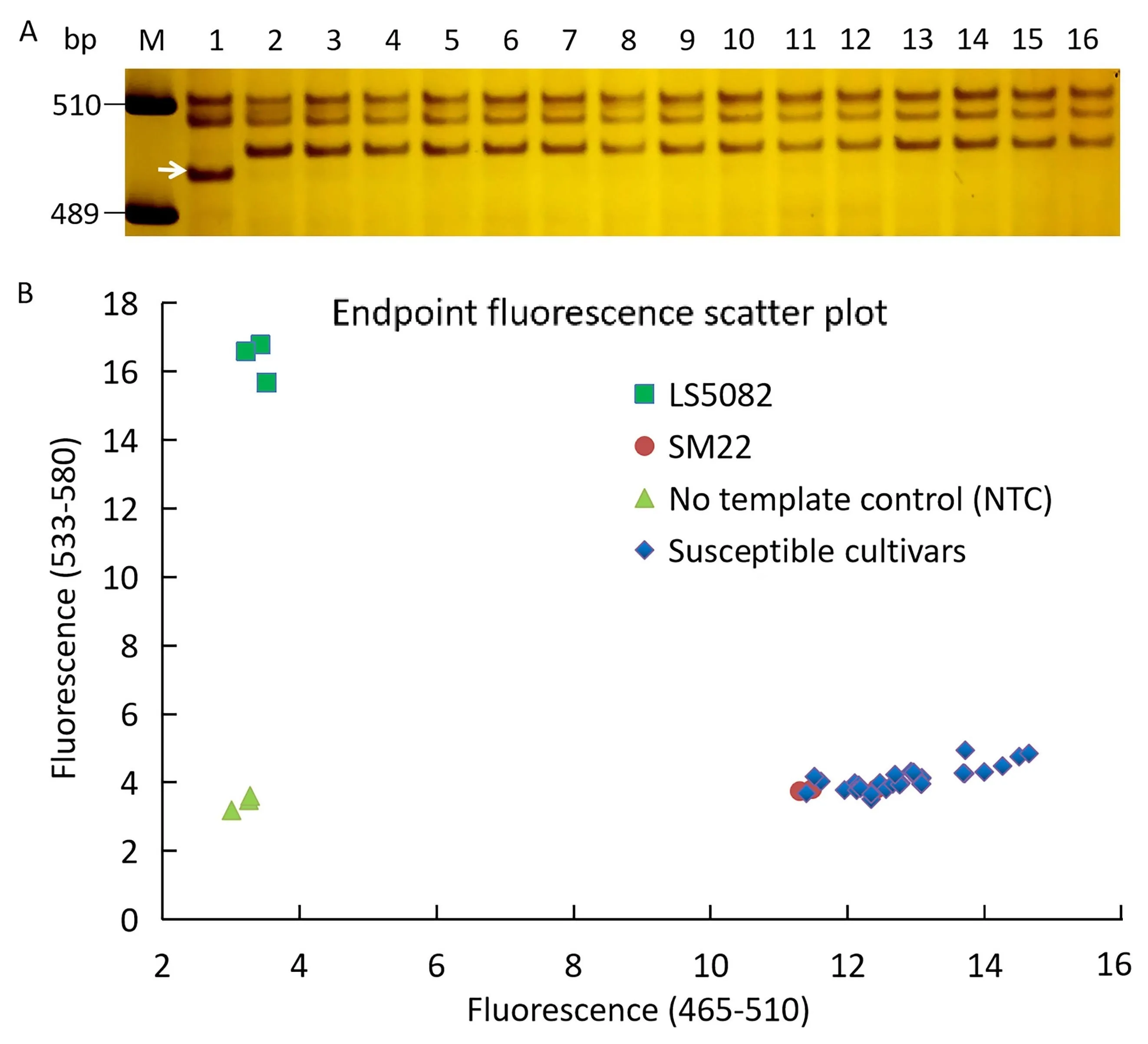
Fig. 6. Amplification patterns of PmLS5082-linked markers YTU19-005 (A) and genotyping results of the PmLS5082-linked kompetitive allele-specific PCR (KASP)markers YTU19-KASP96 (B) for LS5082, Shannong 29 and 15 wheat cultivars/breeding lines susceptible to powdery mildew. Lane M, DNA marker pUC18 Msp I;Lanes 1-2,parents LS5082 and Shannong 29;3,Yannong 187;4,Shannong 1538;5,Zhoumai 27; 6, Zhongyu 1311; 7, Yan 1212; 8, Jimai 229; 9, Zhongyu 9398; 10,Womai 8;11,Danmai 13;12,Daimai 2173;13,Wunong 6;14,Zhengmai 0856;15,Liangxing 619;16,Taimai 1918.Susceptible cultivars in(B)are as follows:Yannong 187, Shannong 1538, Zhoumai 27, Zhongyu 1311, Yan 1212, Jimai 229, Zhongyu 9398, Womai 8, Danmai 13, Daimai 2173, Wunong 6, Zhengmai 0856, Liangxing 619, Taimai 1918.
4. Discussion
LS5082 is an elite wheat breeding line and was maintained in our lab as a powdery mildew resistant germplasm. A dominantRgene,PmLS5082, was genetically identified to confer seedling resistance to 10 of 12 testedBgtisolates. Because Gao 8901, one parent of LS5082, was susceptible to powdery mildew (identified by our lab),PmLS5082is most likely derived from its other parent Xiao 13 that has not been reported to be resistant to powdery mildew.We mappedPmLS5082to chromosome arm 2BL and located it to a 0.7 Mb physical interval (710.3-711.0 Mb). In addition toPmLS5082, 10 otherPmgenes (Pm6,Pm33,Pm51,Pm52,Pm63,Pm64,PmQ,PmKN0816,MlZec1andMlAB10) have been reported on chromosome arm 2BL. The gene donors are from diverse sources, including wheat cultivars, landraces,T. persicumVav.,T.dicoccoides,T.timopheviiZhuk.,andThinopyrum intermediumintrogression lines, suggesting that the chromosome arm 2BL is most likely to be an enrichment region for resistance genes. It also implies that additionalRgenes and complex mechanisms might be located on this chromosome arm.
Based on their chromosome locations and/or their origins,PmLS5082(710.3-711.0 Mb, derived from a wheat breeding line in China)can be distinguished from the knownPmgenes on chromosome 2BL:Pm52(581.0-585.0 Mb)derived from Chinese wheat cultivar Liangxing 99 [40,53];Pm33(779.1-784.3 Mb) derived fromT. persicumVav. [39];MlZec1andMlAB10(both 796.7-780.0 Mb) both derived fromT. dicoccoides[54,55];Pm6(698.3-699.2 Mb) derived fromT. timopheevii2B/2G introgression [37];Pm64(699.2-705.5 Mb) derived from wild emmer [41]; andPmKN0816(700.4-710.3 Mb) derived from Chinese breeding line KN0816 [43]. Due to the fact thatPmLS5082,Pm52andPmKN0816are all derived from Chinese wheat breeding lines and could be easily confused, additional allelism tests betweenPmLS5082andPm52andPmKN0816were carried out to clarify their relationship.Results indicated that they are different genes. Additionally, thePm6interval has severe recombination suppression due to the introgression of the 2G chromosome segment [56]. In contrast,thePmLS5082interval has no significant recombination suppression,and all the markers had normal recombination frequency,further indicating thatPmLS5082is different fromPm6.The physical intervals ofPm51(709.8-739.4 Mb),Pm63(710.3-723.4 Mb) andPmQ(710.7-715.0 Mb) overlap that ofPmLS5082,and hence there was a need for further clarification.Pm51is derived from aTh. intermediumchromosome segment that was introgressed into common wheat [40], suggesting thatPm51have different origins withPmLS5082, and hence can be distinguished fromPmLS5082.Pm63was identified in an Iranian wheat landrace PI 628,024 [57]. None of thePm63-linked markers showed polymorphism between LS5082, Shannong 29 and their derived resistant and susceptible bulks (Table S10), suggesting thatPmLS5082andPm63have different origins and greater genetic diversity in the mapping intervals.PmQis derived from Chinese landrace Qingxinmai,and shows a distinctive recessive inheritance[42], and only onePmQ-linked marker was linked withPmLS5082(Table S10), indicating thatPmLS5082andPmQhave different origins, inherited patterns and greater genetic diversity in the mapping intervals. Furthermore, the resistance spectrum ofPmLS5082was significantly different from all the abovePmgenes. Therefore,we conclude thatPmLS5082is most likely a newPmgene/allele.Even so, to accurately comparePmLS5082with the knownPmgenes on chromosome arm 2BL as highlighted above, more and intensive analysis should be provided in the future,including cloning of these genes. As this interval contains multiple and complexPmgenes, cloning of these genes is necessary to clarify their relationship. Further work is underway to fine mapPmLS5082using larger mapping populations.
To dissect the molecular basis for the powdery mildew resistance and/or analyze candidate gene in LS5082, we also profiled the regulatory and supported genes associated with powdery mildew resistance using BSR-Seq, which is a high efficiency and lowcost means of investigating the overall expression profile of resistance-related genes [35]. AfterBgtinvasion, many DEGs were activated, mainly including those involved in signal transduction mechanisms, defense mechanisms, and carbohydrate transport and metabolism.This result accords with the working model of signal transduction and activation of defense mechanisms:in the case of pathogen invasion, signal transduction mechanisms are expected to be activated in order to transduce the stress signal;subsequently, defense mechanisms are expected to be mobilized to fight the invasion; and both these processes need the support of biosynthesis and metabolism.
To investigate further the key regulatory genes and/or candidate genes involved in the disease-resistance process, the expression profiles of six genes in the candidate interval were analyzed followingBgtinoculation. The functions of these genes can be related to different aspects of the resistance response toBgtinvasion. For example, the LRR receptor-like serine-protein, which is a typical disease resistance protein [58], was expressed continuously in LS5082 following challenge byBgt,and most likely played the most important role in withstanding invasion byBgt. We consider it is the most likely, among the six candidate genes, to bePmLS5082. The G-type lectin S-receptor-like serine-protein and receptor-like serine-protein are powerful gene families related to adversity stress [59,60]. They were induced with high expression in LS5082 at 24 h followingBgtinoculation,indicative of a key role in resistance toBgtinvasion. The zinc finger CCCH domaincontaining protein is an important transcription factor, that can activate downstream resistance genes.The peroxisomal biogenesis factor 6 is related to the functional role of the peroxisome [61],which is to scavenge the active oxygen and hydrogen peroxide produced by theBgtinvasion at the early stage. The abscisic acidinducible protein is a functional protein in the ABA signaling pathway [62], and one of its main functions is regulation of stomatal closure, which may mitigateBgtinvasion to some extent. These data provide an initial attempt at dissecting the resistance pathways and may provide leads towards the improvement of durable resistance.
After dissection of the powdery mildew resistance in a genotype, the next challenge is how to use it effectively in breeding.In resistance breeding,Rgenes are usually transferred into susceptible cultivars to improve their resistance. The advanced breeding line LS5082 showed favorable agronomic performance and broad resistance spectrum to powdery mildew. This makesPmLS5082a valuable resistance gene to broaden the genetic diversity and improve powdery mildew resistance in wheat. To facilitate the transfer ofPmLS5082using MAS, we developed 10 markers that can detectPmLS5082in most of the 27 tested susceptible cultivars(Table S10).It is worth mentioning that we developed two types of breeding markers (gel-based and gel free) forPmLS5082, which meet the needs of different throughput and applicable to different platforms for marker analysis. The gel-based markers can serve as a simple and low-cost selection tool to detect the transfer ofPmLS5082into elite breeding materials. This especially benefits those breeding programs with a basic laboratory setup and makes it possible to use MAS to deployPmLS5082into their cultivars.The KASP marker can be assayed using a SNP genotyping system which can also clearly distinguish homozygotes from heterozygotes in early generation MAS. The KASP marker is gel-free, analyzed by a fluorescence reader [63], and is suitable for those breeding programs with high-throughput marker laboratories. Using these two types of markers, MAS ofPmLS5082is currently underway.F2and F3plants have been selected using both gel-based and gel-free marker alleles. Following selection for agronomic performance in the field,we have now selected two wheat breeding lines with superior agronomic performance and high powdery mildew resistance.
CRediT authorship contribution statement
Liru Wu:Formal analysis, Data curation, Methodology.Tong Zhu:Formal analysis, Methodology.Huagang He:Methodology,Writing - review & editing, Funding acquisition.Xinyou Cao:Methodology,Visualization.Haosheng Li:Methodology,Visualization.Hongxing Xu:Formal analysis, Visualization.Mengshu Jia:Formal analysis.Lipei Zhang:Investigation.Jiancheng Song:Investigation.Ghader Mirzaghaderi:Formal analysis,Conceptualization.Cheng Liu:Conceptualization,Supervision,Funding acquisition.Pengtao Ma:Writing - original draft, Conceptualization,Supervision, Funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We are grateful to Prof. Sishen Li, Shandong Agricultural University, China, for providing the resistant germplasm LS5082.We are grateful to Prof.Paula E.Jameson,University of Canterbury,financially supported by ‘‘Double Hundred” Plan for Foreign Experts in Shandong Province,China,Associate Prof.John Clemens,University of Canterbury,and Dr.Yunfeng Xu,Kansas State University, for critical reviewing and editing of this manuscript. This research was financially supported by the National Natural Science Foundation of China (32072053, 31971874, and 32171990), Taishan Scholars Project(tsqn201812123),Key Research and Development Program of Shandong Province (2020CXGC010805), Open Project Funding of the State Key Laboratory of Crop Stress Adaptation and Improvement(CX1130A0920014),State Key Laboratory of Plant Cell and Chromosome Engineering (PCCE-KF-2019-04), and Iran National Science Foundation (INSF) Grant 99014038.
Appendix A. Supplementary data
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2021.12.008.

- The Crop Journal的其它文章
- Research progress on the divergence and genetic basis of agronomic traits in xian and geng rice
- From model to alfalfa: Gene editing to obtain semidwarf and prostrate growth habits
- The chloroplast-localized protein LTA1 regulates tiller angle and yield of rice
- Genome-wide association study and transcriptome analysis reveal new QTL and candidate genes for nitrogen-deficiency tolerance in rice
- Advances in the functional study of glutamine synthetase in plant abiotic stress tolerance response
- Identification of microRNAs regulating grain filling of rice inferior spikelets in response to moderate soil drying post-anthesis