
Genomic prediction using composite training sets is an effective method for exploiting germplasm conserved in rice gene banks
2022-08-16 09:25SngHeHongynLiuJunhuiZhnYunMengYmeiWngFengWngGuoyouYe
The Crop Journal 2022年4期
Sng He, Hongyn Liu, Junhui Zhn, Yun Meng, Ymei Wng, Feng Wng, Guoyou Ye,b,
a CAAS-IRRI Joint Laboratory for Genomics-Assisted Germplasm Enhancement, Agricultural Genomics Institute in Shenzhen, Chinese Academy of Agricultural Sciences,Shenzhen 518120, Guangdong, China
b Rice Breeding Innovations Platform, International Rice Research Institute, DAPO Box 7777, Metro Manila, Philippines
c Guangdong Provincial Key Laboratory of New Technology in Rice Breeding, Rice Research Institute, Guangdong Academy of Agricultural Sciences, Guangzhou 510640, Guangdong,China
d Hainan Key Laboratory for Sustainable Utilization of Tropical Bioresource, College of Tropical Crops, Hainan University, Haikou 570228, Hainan, China
Keywords:Genomic prediction Composite training set Rice germplasm Gene bank Reliability criterion
A B S T R A C T Germplasm conserved in gene banks is underutilized, owing mainly to the cost of characterization.Genomic prediction can be applied to predict the genetic merit of germplasm. Germplasm utilization could be greatly accelerated if prediction accuracy were sufficiently high with a training population of practical size.Large-scale resequencing projects in rice have generated high quality genome-wide variation information for many diverse accessions, making it possible to investigate the potential of genomic prediction in rice germplasm management and exploitation. We phenotyped six traits in nearly 2000 indica (XI) and japonica (GJ) accessions from the Rice 3K project and investigated different scenarios for forming training populations. A composite core training set was considered in two levels which targets used for prediction of subpopulations within subspecies or prediction across subspecies. Composite training sets incorporating 400 or 200 accessions from either subpopulation of XI or GJ showed satisfactory prediction accuracy. A composite training set of 600 XI and GJ accessions showed sufficiently high prediction accuracy for both XI and GJ subspecies. Comparable or even higher prediction accuracy was observed for the composite training set than for the corresponding homogeneous training sets comprising accessions only of specific subpopulations of XI or GJ(within-subspecies level)or pure XI or GJ accessions (across-subspecies level) that were included in the composite training set. Validation using an independent population of 281 rice cultivars supported the predictive ability of the composite training set. Reliability, which reflects the robustness of a training set, was markedly higher for the composite training set than for the corresponding homogeneous training sets. A core training set formed from diverse accessions could accurately predict the genetic merit of rice germplasm.
1. Introduction
In rice, a staple food for over half of the human population [1],production and selection gain have rocketed in recent decades,thanks to the Green Revolution [2-4]. However, these gains have been accompanied by a narrowing of genetic diversity, impeding long-term selection gain in rice breeding. Rice gene banks around the world conserve abundant rice germplasm, with that of the International Rice Research Institute (IRRI) holding more than 140,000 accessions. These gene banks provide rich genetic resources to reinforce the genetic diversity consumed in breeding,but remain underutilized because constraints of budgets and logistics prevent extensive phenotyping of germplasm.
Genomic prediction can efficiently genetically characterize germplasm without the need for comprehensive phenotyping, as described by Yu et al. [5]. The accuracy of genomic prediction depends strongly on the robustness of the training set.As relatedness is the major factor driving genomic prediction accuracy[6-8],a prerequisite of the training set is that it should be genetically related to the genotypes to be predicted. For this reason, genomic prediction is implemented within specific subspecies or subpopulations. But because germplasm preserved in gene banks is highly diverse, compiling a unique training set for each specific subspecies or subpopulation is impractically expensive. A composite training set consisting of accessions from each subspecies or subpopulation offers potential for comprehensive prediction, as it is related to each subspecies or subpopulation. The utility of such a set has been substantiated in dairy cattle breeding. Hayes et al.[9] and Erbe et al. [10] showed that including Holstein cattle in a training set for a Jersey population increased the genomic predictability of Jersey cattle in comparison with the pure Jersey training set.Hoze et al.[11]demonstrated that a multi-bred dairy cattle training population could improve prediction for the breed with a small number of reference animals included in the composite training set.Guo et al.[12]investigated the potential of a composite training set in rice and found the prediction accuracy using a composite training set of XI, GJ-trp, and GJ-tmp (rice subpopulations) to be comparable to that based on homogeneous training sets.However,as they used only around 400 accessions,the results await validation in a larger and more diverse population.
The Rice 3K project [13] collected more than 3000 diverse accessions from the gene banks of IRRI and China. They include all major known rice subspecies and ecotypes and provide abundant genetic resources for developing composite training sets.Ideally, the composite training set can be organized among subspecies, which enables prediction of any subspecies contained in the training set. However, the two rice subspecies,indica(XI)andjaponica(GJ), are genetically so distant that they are adapted to very different ecological conditions. Merging accessions from these subspecies may greatly reduce the overall relatedness between the training set and the accessions to be predicted,impairing prediction in either subspecies [14,15]. The expansion of the training set conferred by combining accessions from different subspecies may not compensate for the sharp decrease in relatedness of the training set to individual subspecies. Compiling a composite training set across subpopulations within a specific subspecies is more promising,as relatedness among subpopulations is much greater.Still,the potential of a composite training set of both levels: across subspecies and across subpopulations within subspecies,merits investigation.In addition,the utility of a composite training set also depends on the trait of interest. For some traits measured at early phases of plant growth such as the seedling stage, different subspecies are not separately measured and an across-subspecies composite training set can be organized to predict accessions of each subspecies.For traits measured at maturity such as grain yield, different subspecies are normally respectively evaluated.Thus,a composite training set organized across subpopulations within specific subspecies is useful.
To investigate the potential of a composite training set for genomic prediction for germplasm utilization in rice,we measured four early traits and two maturity-stage traits for 1999 accessions from the Rice 3K project. An independent population consisting of 281 cultivars was used to validate the robustness of the composite training set.Our experimental aims were to investigate 1)whether the composite training set organized across subpopulations within XI or GJ showed sufficiently high prediction accuracy for accessions from each subpopulation of XI or GJ, and 2) whether the use of a composite training set built across subspecies XI and GJ is adequate for an accurate prediction of germplasm of XI or GJ.
2. Materials and methods
2.1. Rice materials
Subsets of 1338 XI and 661 GJ accessions from the Rice 3K project were sampled. Following Wang et al. [13], XI accessions were divided into subpopulations XI-1A, XI-1B, XI-2, XI-3, and XI-adm,based mainly on their geographic origins. In view of their genetic similarity and subpopulation size,XI-1A and XI-1B were combined as XI-1. GJ accessions were sampled from subpopulations GJ-trp,GJ-sbtrp, and GJ-tmp [13]. GJ-sbtrp and GJ-tmp were merged into a more general subpopulation, GJ-tmp, following classical taxonomy [16,17]. The size of each subpopulation is shown in Table 1.All accessions were phenotyped for six traits: mesocotyl length,coleoptile length, shoot length, germination rate, normalized difference vegetation index (NDVI), and straw weight. A population of 281 cultivars from south Asia, southeast Asia, and China and phenotyped for mesocotyl length was used for independent validation.
2.2. Phenotyping
To evaluate the variation in mesocotyl length(the distance from the basal part of the seminal root to the coleoptile node),coleoptile length, and shoot length, the phenotyping procedure established by Liu et al. [18] was used. The detailed phenotyping method was as follows: 15 good-quality seeds from each accession were sown at a depth of 6 cm in plant substrate contained in plastic trays, in each replicate. Each plastic tray contained 50 holes, each with 9.5 cm depth,4.5 cm top diameter,and 2.1 cm bottom diameter. After sowing, the plastic tray was kept in a container with 3 cm deep soil, and the system was maintained in a 30 °C dark incubator. Soil in each pallet was kept water-saturated for seed emergence and seedling growth. Emergence rate was recorded every day, until the emergence rate of any accession reached 100%.The reading at the last day was recorded for calculating germination rate.Three days after that,seedlings were carefully excavated and washed for measurement of mesocotyl length,coleoptile length, and shoot length using Image J (https://imagej.en.softonic.com/). A randomized complete block design with two replicates was adopted as a layout for the experiment. The phenotypic values of mesocotyl, coleoptile, and shoot length used for genomic prediction were the means of two replications.
To measure NDVI,a field trial was conducted at the experiment station of the Agricultural Genomics Institute in Shenzhen, China.A row-column design with two complete replicates was used.The methodology for NDVI measurement followed Condorelli et al. [19]. NDVI was measured after eighty days of sowing using an unmanned aerial vehicle (UAV) platform. UAV-based NDVI was extracted from georeferenced orthomosaic GeoTIFFs generated from imagery captured from autopiloted flights of a Parrot Sequoia (Parrot, Paris, France) multi-spectral camera carried on a fixed-wing aircraft (Dajiang-Spirit, Shenzhen, Guangdong, China).Table S1 shows the features of the multispectral camera in terms of band centers and bandwidths. Flights were conducted at 30 m above ground level, resulting in ground sampling resolution of~3 cm/pixel for the Sequoia.Mission planning was performed with universal ground control software for the Sequoia camera (Mica-Sense, Seattle, WA, USA). All flights were planned for 80% image overlap along flight corridors. Pix4D Mapper Pro desktop software(Pix4D SA, Switzerland, http://pix4d.com) was used to generate orthomosaics for each camera band. Six to eight ground control points (GCP) geolocated with Real Time Kinematic (RTK) survey precision was used to georeference the orthomosaics. Camera images were calibrated using the manufacturer’s reflectance panels, which were imaged at the beginning of each flight. The Pix4D processing options were essentially the same as those of Pix4D’s‘‘Ag Multispectral” template version 4.1.10, except that GeoTIFF tiles were merged to create the NDVI orthomosaic.Plot-level NDVI means from UAV’s were calculated in Quantum Geographic Information System (QGIS) software version 2.18.3 (QGIS, US, http://www.qgis.org). Shape files containing annotated single-plot polygons were generated using R[20].Shape files with GCPs as features(points) were also employed based on RTK survey-grade measuring devices.For all flights,the GeoTIFF with the NDVI orthomosaicfrom Pix4D was combined with the plot polygon and GCP shape files in a single QGIS project. Confirmation of proper geolocations of the Pix4D orthomosaics was achieved by visually confirming alignment of the visible GCPs with the corresponding points in the feature shape file. NDVI plot means were generated with the Zonal Statistics function in QGIS.To eliminate bias from bird damage in dry-matter production of early-maturity genotypes, only rice straw was harvested to represent aboveground biomass. By hand threshing, all spikelets were separated from the rachis, and the remaining parts were oven-dried at 70 °C to constant weight.
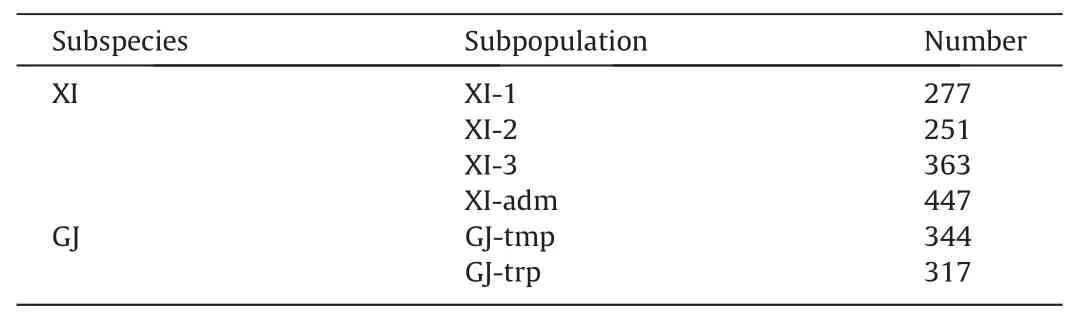
Table 1The number of accessions in each subpopulation of indica (XI) and japonica (GJ).
The lme4 package [21] was used to conduct a spatial adjustment analysis of the raw NDVI plot data using a mixed model.Row and column were treated as random effects and a moving mean of variable size was used.
2.3. Genomic data
The resequencing and variant-calling protocols of the 1338 XI and 661 GJ accessions from the Rice 3K project were described by Wang et al. [13]. The original genotypic data were retrieved from https://aws.amazon.com/public-data-sets/3000-rice-genome/. In total, 26,829,979 bi-allelic single nucleotide polymorphisms (SNPs) were available. Further quality control for SNPs was performed by (1) removing SNPs with minor allele frequency(MAF) <0.05; (2) removing SNPs with call rate <90%; (3) filtering SNPs using the linkage disequilibrium (LD) pruning procedure in PLINK [22] with a window size of 300 SNPs, shifting step of 50 SNPs, andr2threshold of 0.1. Finally, 24,887 high quality SNPs were available for the 1999 genotypes. The resequencing and variant-calling procedure of the independent population of 281 cultivars was as described by Wang et al. [13] except that the sequencing depth was 50×.Finally,11,178,242 biallelic SNPs were identified, of which 18,770 were common to the 1999 3K accessions and the independent population.Relatedness between accessions was inspected using a cluster analysis based on the 18,770 common SNPs.
To investigate the potential of a composite training set for subpopulations within subspecies,the genotypic data were readjusted for XI and GJ populations separately.Quality control was applied as described for the full set of 1999 accessions. Finally, 53,416 and 14,878 SNPs were available for the XI and GJ populations,respectively.
2.4. Genomic prediction models


The two BLUP models were implemented in R [20] using the BGLR package [26]. The Bayesian approaches were fitted with GCTB software developed by Zeng et al. [27]. Genomic prediction accuracy was calculated as the Pearson correlation coefficient between adjusted phenotypic values and genomic predicted genetic values.
2.5. Genomic heritability
Genomic heritability of each trait was independently estimated in the full population of 1999 genotypes and in the two subspecies populations XI and GJ using the formulabased on the GBLUP model.GCTA software[28]was used to compute the genomic relationship matrix and fit the GBLUP model.
2.6. Composite training set designs and prediction scenarios
The composite training set was organized in two levels:withinsubspecies and across-subspecies. For the within-subspecies level,the composite training set comprised accessions from each subpopulation of the XI, and GJ. Predictive ability was validated in each subpopulation of XI or GJ. To allow investigating the effect of training set size, 5%-80% accessions from each subpopulation were stochastically sampled to compose the composite training set, while the remaining 95%-20% accessions formed test sets.Genomic prediction accuracy was separately evaluated in each subpopulation. Random sampling of accessions to compile the composite training set was repeated 100 times. For comparison,samples from a specific subpopulation, termed as a homogeneous training set, were also used to predict the corresponding test set belonging to the same subpopulation.
For the across-subspecies level, both XI and GJ samples in each repeat of the random sampling with sampling ratios of 5%-80% in the within-subspecies-level composite training set design described above were combined as the composite training sets,while the remaining 95%-20%XI accessions formed the XI test sets.The GJ test sets were constructed likewise. Genomic prediction accuracy was separately computed in the XI and GJ test sets. The XI and GJ test sets were also predicted using the sampled accessions of only their own subspecies, which were denoted as homogeneous training sets. The composite training set designs for both within-subspecies and across-subspecies levels are shown in Fig. S1.
Genomic predictive ability of the across-subspecies level composite training set was further validated in the independent population of 281 cultivars. These cultivars were first genetically clustered with reference to the Rice 3K accessions. Genomic prediction accuracy was separately evaluated in each clustered set.
2.7. Reliability criterion
As reliability criterion theoretically describes the correlation between predicted and true genetic values [29-31] and is also indicative of genomic prediction accuracy [32], we compared reliability estimates derived from the composite and corresponding homogeneous training sets. The reliability of each accession in the test set was estimated with the GBLUP model and was formulated aswhere PEViis the prediction error variance [29] of theithgenotype. GBLUP was implemented using functionmmerin the R package sommer[33]andPEVcan be directly extracted from columnPevUin the output of functionmmer.
3. Results
3.1. Range and coefficient of variation (CV) of phenotypic values,genomic heritability, and population classification of the 281 independent cultivars
The ranges of phenotypic values of the six traits under study are shown in Table 2.Among the six traits,the CVs of NDVI and mesocotyl length measured among all 1999 XI and GJ accessions or separately in the XI and GJ sets were respectively smallest and largest,at approximately 0.1 and 0.6.Genomic heritability estimates of the six traits studied were overall highest in XI,and were similar in full population and GJ (Table 2). Of the 281 cultivars of the independent population, 191 were genetically close to XI and the remaining 90 were close to GJ (Fig. S2).
3.2. Within-subspecies genomic prediction accuracy and reliability
Genomic prediction accuracies using a composite training set organized among subpopulations within subspecies XI or GJ increased overall with the size of the training set for all traits,prediction models, and validation subpopulations (Figs. 1, 2, S3-S12).In most cases, using composite training sets resulted in insignificantly different or significantly higher prediction accuracies relative to the corresponding homogeneous training sets incorporating accessions only from the same subpopulation as the accessions in the test sets.The number of cases in which using a composite training set led to significantly higher prediction accuracies than using the corresponding homogeneous training set was greater for the Bayesian models than for the BLUP models. For XI,when the composite training set size was small (5%-10%, corresponding to 67-134 accessions), the prediction accuracies of BayesR were overall markedly lower than those of other prediction models for all traits considered (Figs. 1, S3-S7). The prediction accuracies of different subpopulations using the composite training set differed,except for mesocotyl and coleoptile length.In contrast, prediction accuracies of the two GJ subpopulations differed among all traits studied (Figs. 2, S8-S12).
The prediction accuracies of the composite training sets reached their plateau approximately when 30%of the accessions from each subpopulation of XI or GJ, or about 400 accessions of XI or 200 of GJ, were included in the composite training set (Figs. 1, 2, S3-S12). For XI, the composite training set comprising approximately 400 XI accessions (30%) from each subpopulation did not result in significantly lower prediction accuracies than the homogeneous training sets incorporating more than 30% accessions of a specific XI subpopulation (Table S2). The number of cases in which the composite training set containing 400 XI accessions (30%) was not inferior to larger composite training sets using the Bayesian models was greater than that using the BLUP models (Table S2).For GJ, the composite training set comprising approximately 200 GJ accessions (30%) from each subpopulation did not yield significantly lower prediction accuracy than the homogeneous training sets comprising even more than 200 accessions (70%-80% for GJtrp and 60%-80%for GJ-tmp,corresponding to 222-254 accessions for GJ-trp and 206-275 accessions for GJ-tmp) (Table S3). The Bayesian approaches performed better than the BLUP methods(Table S3).
For all the investigated training set sizes, reliability estimates achieved by the composite training sets were globally higher than those by the homogeneous training sets (Figs. 3, 4). The variation among reliability estimates in the XI subpopulations was overall smaller than that in the GJ subpopulations (Figs. 3, 4). Comparing different subpopulations,the superiority of the composite training set over the homogeneous training sets in reliability estimation varied with the trait (Figs. S13, S14).
3.3. Across-subspecies genomic prediction accuracy and reliability
Similarly to the trend observed in the within-subspecies level,the prediction accuracies using the across-subspecies composite training set increased with training set size for all traits, models,and validation subspecies (Figs. 5, S15-S19). In most cases, using a composite training set resulted in insignificantly different or significantly higher prediction accuracies relative to the corresponding homogeneous training sets incorporating germplasm only from the same subspecies as the germplasm in the test sets(Figs.5,S15-S19). However, for straw weight, the prediction accuracies of XI accessions using the composite training set were mostly significantly lower than those using the corresponding homogeneous training set, except for the EGBLUP model (Fig. S19). Prediction accuracy of XI was overall higher than that of GJ accessions for coleoptile length and shoot length (Figs. S15, S16), whereas for the other four traits the GJ displayed generally higher prediction accuracy than XI accessions (Figs. 5, S17-S19). This difference could not be explained by the differences in the estimates of heritability for XI and GJ(Table 2).For coleoptile length,the prediction accuracies of GJ accessions when the composite training set size was small (5%-10%, corresponding to 100-200 accessions) were markedly lower for the BayesR model than for the other prediction methods (Fig. S15).
The prediction accuracies using the composite training set reached their plateau when 30% accessions from XI and GJ, of the full 600 accessions, were included in the composite training set(Figs.5,S15-S19).Prediction accuracies using the composite training set comprising 600 XI and GJ accessions were not significantly lower than those of the homogeneous training sets incorporating more than 30% accessions (40%-80%, corresponding to 535-1070 XI genotypes and 264-529 GJ genotypes), especially for the GJ homogeneous training sets when the BayesB model was used(Table S4).
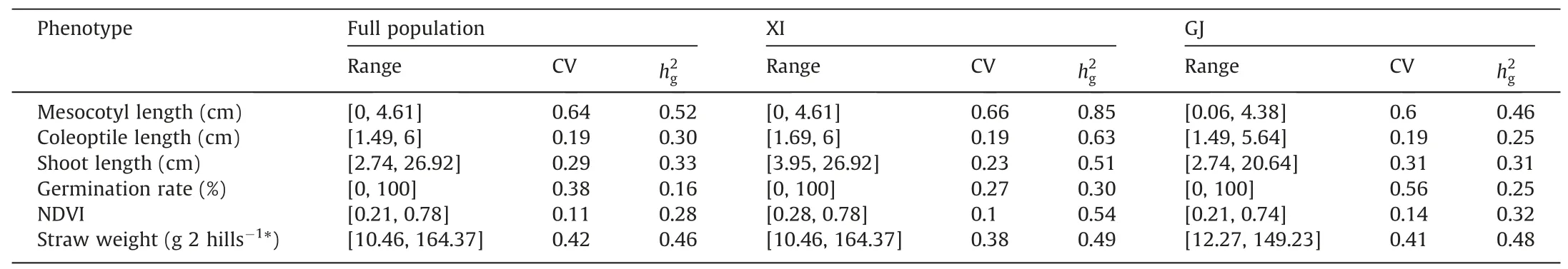
Table 2Range and coefficient of variation(CV)of phenotypic values,and genomic heritabilities(?)of traits,estimated from the full population or separately from indica(XI)and japonica(GJ) sets.
For each training set size,reliability was higher for the composite training set than for the homogeneous training sets (Fig. S20).The use of a composite training set seemed particularly beneficial for GJ. For instance, the composite training set consisting of 600 XI and GJ accessions (30% of the full accessions)(Fig. S21) showed markedly higher reliability estimates than the GJ homogenous training set for all of the traits.
3.4. Validation of genomic predictive ability of the composite training set in an independent population
Genomic prediction accuracy of mesocotyl length of XI and GJ in the independent population using the composite training setcomposed of 600 XI and GJ accessions(30%)from the Rice 3K project was slightly lower than that observed in validation within the 3K population, decreasing from approximately 0.6 to 0.5 (Figs. 5,6). Prediction accuracies of the GJ group were overall higher than those of XI accessions,in accord with that within the 3K population(Figs. 5, 6). The Bayesian approaches performed more poorly than the BLUP models with respect to both overall prediction accuracy and variation of prediction accuracy for both XI and GJ groups(Fig. 6).
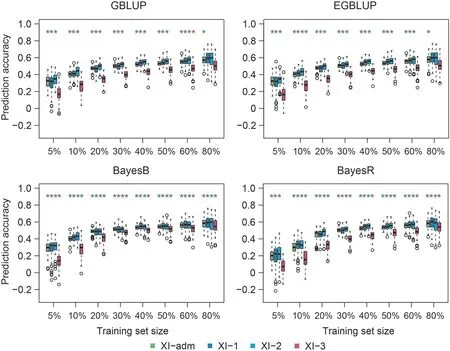
Fig. 1. Genomic prediction accuracies of mesocotyl length in subpopulations of indica (XI) using four prediction approaches based on a composite training set consisting of approximately 70(5%)to 1070(80%)accessions from each subpopulation of XI.Green and red asterisks indicate that the prediction accuracies using the composite training set were respectively higher and lower(P <0.05, t-test) than the accuracies using a homogeneous training set comprising accessions in the composite training set from the same subpopulation as the test set. Training set size is displayed as the ratios of sampling accessions from each subpopulation.
4. Discussion
4.1. A composite training set permits comprehensive and accurate prediction
In comparison with the preceding study of Guo et al. [12], ours used much more diverse accessions and demonstrated that a composite training set composed of about 600 XI and GJ accessions satisfactorily predicted each of the two subspecies. However, the prediction accuracy was not satisfactory when the composite set was reduced to the size used by Guo et al.[12].The germplasm collection of the Rice 3K project has high diversity and appropriately represents the diversity in the world largest rice gene bank [13].For this reason,the composite training set compiled in the present study could be used for predicting other XI and GJ accessions conserved in the IRRI gene bank or newly collected. Compared to the validation within the Rice 3K population, only a slight reduction of prediction accuracy was observed in the independent population consisting of modern cultivars, supporting the robustness of the composite training set (Fig. 6). The validation in the independent population could also indicate that the training set composed of landraces and primitive cultivars could also be used to predict the genetic merits of modern rice cultivars.Still,other independent validation populations with phenotypes of more traits are needed to corroborate the robustness of the composite training set.For the traits studied,which showed a wide range of heritability estimates,the composite training set did not lead to inferior prediction accuracies relative to the homogeneous training sets (Figs. 1, 2, 5,S3-S12, S15-S19), implying that the composite training set could be used for predicting various traits.Comparing the predictabilities when the composite training set was organized within and across subspecies, the within-subspecies composite training set did not provide higher prediction accuracies across different traits than the across-subspecies composite training set. Theoretically, a within-subspecies composite training set would lead to higher prediction accuracies than an across-subspecies training set, because the relatedness between subpopulations within a specific subspecies is closer than that between subspecies.However,considering another critical factor affecting prediction accuracy, namely training set size[34-36],the size of the within-subspecies composite training set was much smaller than that of the acrosssubspecies set. Thus, the benefit of genetic closeness between the within-subspecies composite training set and each included subpopulation would be counterbalanced by its small size.
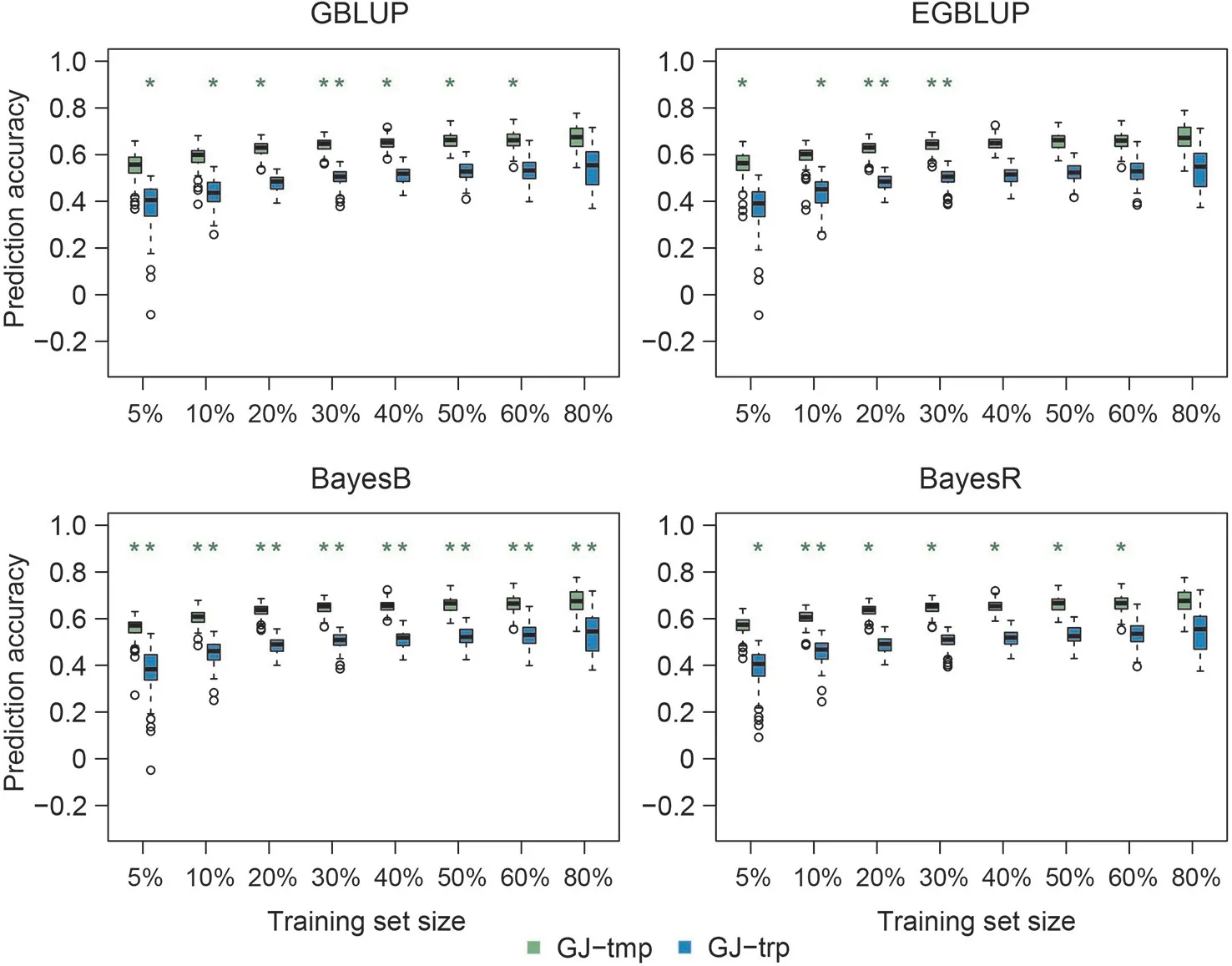
Fig.2. Genomic prediction accuracies of mesocotyl length in subpopulations of japonica(GJ)using four prediction approaches based on a composite training set consisting of approximately 30(5%)to 530(80%)accessions from each subpopulation of GJ.Green and red asterisks indicate that the prediction accuracies using the composite training set were respectively higher and lower(P <0.05,t-test)than the accuracies using a homogeneous training set comprising accessions in the composite training set from the same subpopulation as the test set. Training set size is displayed as the ratios of sampling accessions from each subpopulation.
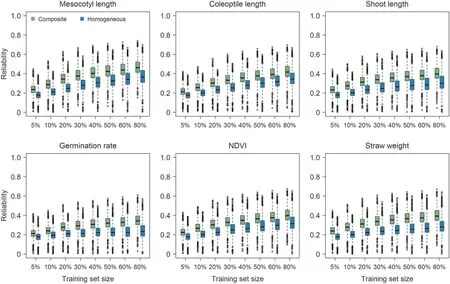
Fig. 3. Reliability estimates of accessions in test sets for all traits based on GBLUP model using a composite training set consisting of approximately 70 (5%) to 1070 (80%)accessions from each subpopulation of indica(XI)and homogeneous training sets comprising accessions in the composite training set from the same subpopulation as the test set. Training set size is displayed as the ratios of sampling accessions from each subpopulation.
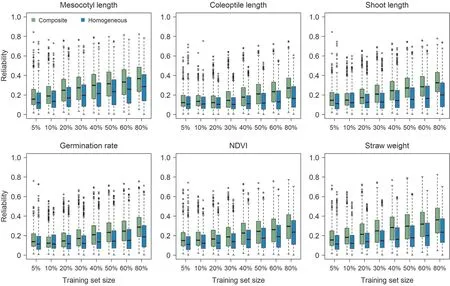
Fig. 4. Reliability estimates of accessions in test sets for all traits based on GBLUP model using a composite training set consisting of approximately 30 (5%) to 530 (80%)accessions from each subpopulation of japonica(GJ)and homogeneous training sets comprising accessions in the composite training set from the same subpopulation as the test set. Training set size is displayed as the ratios of sampling accessions from each subpopulation.
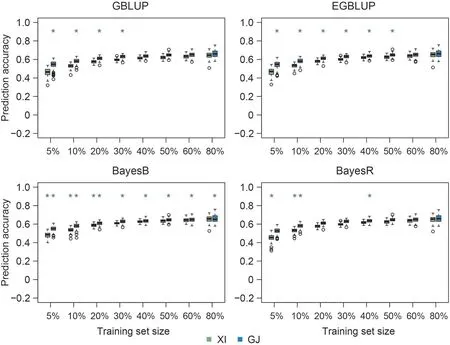
Fig. 5. Genomic prediction accuracies of mesocotyl length in indica (XI) and japonica (GJ) populations using four prediction approaches based on a composite training set consisting of 100(5%)to 1600(80%)accessions from both indica(XI)and japonica(GJ)populations.Green and red asterisks indicate that the prediction accuracies of XI or GJ accessions using composite training set were respectively higher and lower (P <0.05, t-test) than the accuracies using a homogeneous training set comprising only XI or GJ accessions involved in the composite training set. Training set size is displayed as the ratios of sampling accessions from each subspecies.
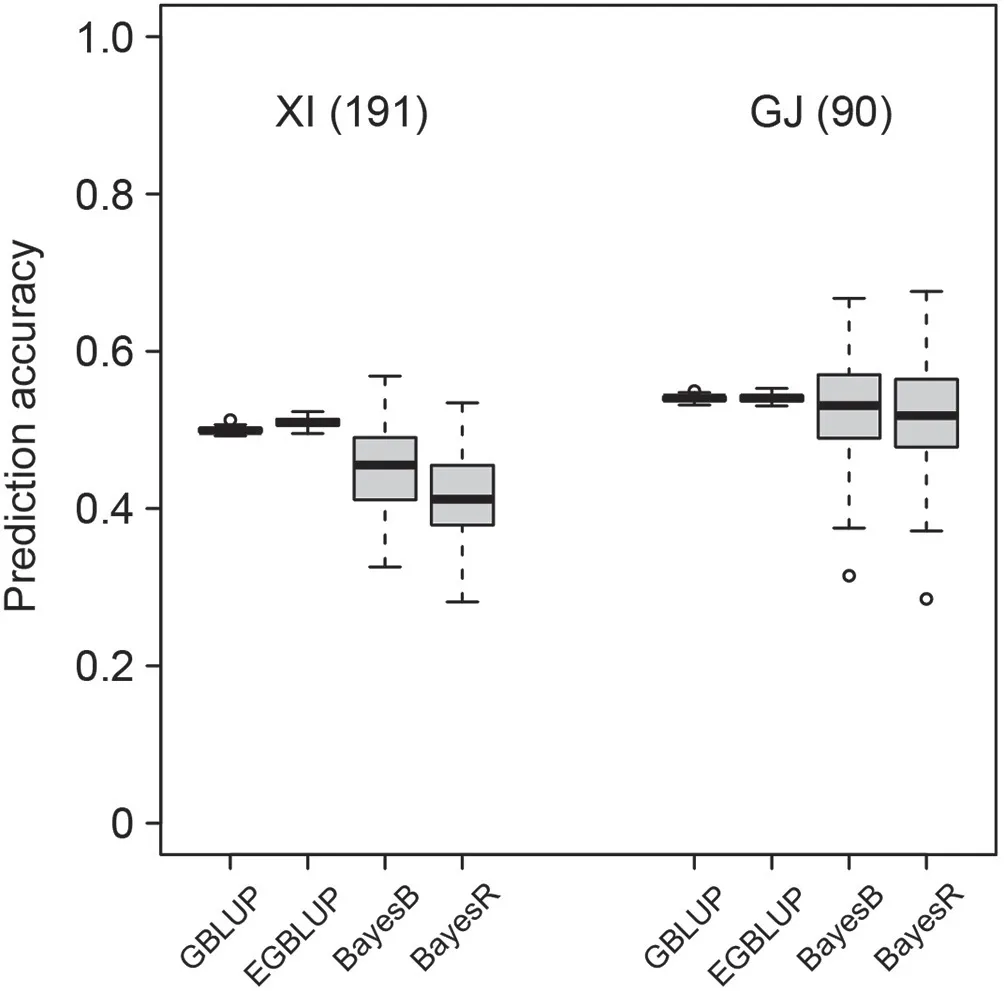
Fig. 6. Genomic prediction accuracies of mesocotyl length in indica (XI) and japonica(GJ)groups of the independent population based on four prediction models using composite training sets consisting of 600(30%)XI and GJ accessions from the Rice 3K project.Numbers in parentheses indicate numbers of cultivars belonging to the XI and GJ groups.
4.2.A composite training set consisting of a few hundred accessions is sufficient
The composite training set with a few hundred accessions is sufficient as its prediction accuracy reached the plateau and was insignificantly different or significantly higher than those of corresponding homogeneous training sets (Figs. 1, 2, 5, S3-S12, S15-S19). Moreover, the prediction accuracy of composite training set was even not significantly inferior to the homogeneous training sets that included more accessions of specific subpopulations/subspecies (Table S2-S4). In particular, using the composite training set established for the GJ showed no significant reduction in prediction accuracies for each of the subpopulations relative to the homogeneous training sets containing more than 200 accessions(Table S3), reducing the necessity of organizing homogeneous training sets specifically for each GJ subpopulation. The variation of prediction accuracies among different samplings of reference accessions to constitute the composite training set was small for both of the within-subspecies level (400 and 200 for XI and GJ,respectively, Figs. 1, 2, S3-S12) and across-subspecies level (600 XI and GJ accessions,Fig.5,S15-S19),indicating that the predictive ability of the composite training set was stable and that the training set could be formed in practice by random sampling.Although the constitution of the composite training set could be further optimized by selecting representative accessions [5,30], the benefit from such optimization is likely to be small.
4.3. BLUP models are recommended in across-population prediction
There was negligible difference in prediction accuracies between the BLUP models (GBLUP and EGBLUP) and the Bayesian approaches (BayesB and BayesR) in different prediction scenarios within the 3K population when composite training sets were used(Figs. 1, 2, 5, S3-S12, S15-S19). However, with respect to the prediction accuracies of mesocotyl length using different genomic prediction models in the independent population based on the composite training set composed by the 3K project accessions,the BLUP models performed much more stably than the Bayesian models in the two sets classified as XI and GJ (Fig. 6). This finding could be attributed to the difference between BLUP and Bayesian models in hypotheses about the distribution of marker effects. In BLUP models, marker effects are assumed to be independently and identically distributed [24,37], in accordance with the infinitesimal model underlying quantitative traits[38,39].In Bayesian models,varied distributions of marker effects are set with the assumption that some markers are linked with major genes or large-effect quantitative trait loci (QTL) [24,40]. For this reason,the performance of Bayesian approaches is highly influenced by the existence of major genes or QTL and the linkage disequilibrium(LD)between markers and genes/QTL[36,41].We used LD of pairs of SNPs as surrogate of LD between SNPs and genes or QTL and found the that the LD of SNPs across the genome separately estimated in the 3K population and independent population varied greatly (Fig. S22). When a population is highly diverse it is highly likely that in different subpopulations the markers in high LD with QTL are different. This reduces the benefits from assuming different marker effect distributions by the Bayesian approaches,which is more appropriate for more homogeneous populations, or even makes the assumption a disadvantage.In contrast,the BLUP methods straightforwardly estimate the overall genetic value of accessions based on a genomic relationship matrix, circumventing the problem raised by the inconsistency of the LD extent of markers among populations. For this reason, we recommend the use of BLUP models for independent populations prediction if the proposed composite training set is used.
4.4. The composite training set improves prediction reliability of genotypes to be predicted
Theoretically,the prediction reliability of a specific accession is the squared correlation between predicted and true genetic value[31,42]. The method used in our study to calculate heritability is based on the prediction error variance, which can be directly derived from the inverse of the coefficient matrix of a mixed model equation[29].The coefficient matrix contains the genetic relationship matrix of accessions and variance component estimates of random effects, e.g., genetic effect and residual. These two factors,the genetic relationship matrix and variance component estimates,are associated with relatedness and training set size,which are the two major factors driving genomic prediction accuracy[7,8,11,34,43]. Relatedness and training set size are intrinsically in a counterbalancing relationship,as integrating more genetically distant accessions into the training set would expand the training set size but lower the overall relatedness between the training set and the genotypes to be predicted. Still, the reliability criterion could integrally consider these two factors and afford an overall evaluation of robustness of a training set. Several studies[42,44,45]have used reliability or reliability-associated parameters to optimize training set design.
Our study demonstrated that a composite training set,whether organized in the level of within-subspecies or across-subspecies,could lead overall to higher reliabilities than the corresponding homogeneous training sets(Figs.3,4,S13,S14,S20,S21).This finding is in line with that of Hayes et al.[9]showing that a composite training set incorporating both Holstein and Jersey cattle could boost the reliability estimates of both Holstein and Jersey cattle compared to a single-breed training set, and a simulation study of De Roos et al. [31] showing that combining populations could increase the reliabilities of both populations especially when the populations were not diverged and a high density of markers was available. In our study, the differences between reliabilities of small population(i.e.,GJ)accessions estimated based on the composite and homogeneous training sets were markedly larger than the differences for large population (i.e., XI) accessions (Fig. S21). For GJ accessions the number of cases in which the composite training set resulted in significantly higher prediction accuracies than the homogeneous training set were greater than that for XI(Figs.5,S15-S19).Similar observations were also made in dairy cattle studies by Hayes et al.[9],Erbe et al.[10],and Hoze et al.[11].This finding implies that relative to the benefit brought by the expansion of training set size,the negative impact on overall relatedness incurred by integrating genetically distant reference genotypes would be marginal if the genetically close reference genotypes, i.e., accessions belonging to the same subpopulation or subspecies as the test set accessions respectively in the level of within- or across-subspecies, have been included in the composite training set.
5. Conclusions
In approximately 2000 XI and GJ accessions from the Rice 3K project, a composite training set containing 400 or 200 accessions from XI or GJ showed satisfactory genomic prediction accuracy for accessions from each subpopulation of XI or GJ.A composite training set containing 600 XI and GJ accessions showed adequately high prediction accuracies for the XI and GJ accessions.A composite training set afforded prediction accuracy comparable to or higher than those of homogeneous training sets consisting of accessions from specific subpopulations of XI or GJ when organized within XI or GJ populations, or pure XI or GJ accessions when established across XI and GJ populations.Validation using an independent population of 281 rice cultivars phenotyped for mesocotyl length supported the predictive ability of the composite training set.The superiority of the composite training set over the homogeneous training sets was attributed to increased reliability, revealing the robustness of a training set.
CRediT authorship contribution statement
Sang He:Writing-original draft,Investigation,Formal analysis,Conceptualization, Methodology.Hongyan Liu:Writing - original draft,Resources,Data curation.Junhui Zhan:Resources,Data curation.Yun Meng:Resources, Data curation.Yamei Wang:Resources,Data curation.Feng Wang:Conceptualization,Methodology.Guoyou Ye:Writing - review & editing, Resources, Data curation, Conceptualization, Methodology.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This project was funded by National Key Research and Development Program of China(2020YFE0202300)and International Postdoctoral Exchange Fellowship Program (Talent-Introduction Program) in 2020.
Appendix A. Supplementary data
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2021.11.011.

- The Crop Journal的其它文章
- Research progress on the divergence and genetic basis of agronomic traits in xian and geng rice
- From model to alfalfa: Gene editing to obtain semidwarf and prostrate growth habits
- The chloroplast-localized protein LTA1 regulates tiller angle and yield of rice
- Genome-wide association study and transcriptome analysis reveal new QTL and candidate genes for nitrogen-deficiency tolerance in rice
- Advances in the functional study of glutamine synthetase in plant abiotic stress tolerance response
- Identification of microRNAs regulating grain filling of rice inferior spikelets in response to moderate soil drying post-anthesis