
Gut microbiota, inflammatory bowel disease and colorectal cancer
2022-08-13 07:04AnaElisaValenciseQuaglioThaisGagnoGrilloEllenCristinaSouzaDeOliveiraLuizClaudioDiStasiLigiaYukieSassaki
World Journal of Gastroenterology 2022年30期
Ana Elisa Valencise Quaglio, Thais Gagno Grillo,Ellen Cristina Souza De Oliveira, Luiz Claudio Di Stasi, LigiaYukie Sassaki
Abstract The gut microbiota is a complex community of microorganisms that inhabit the digestive tracts of humans, living in symbiosis with the host. Dysbiosis, characterized by an imbalance between the beneficial and opportunistic gut microbiota,is associated with several gastrointestinal disorders, such as irritable bowel syndrome (IBS); inflammatory bowel disease (IBD), represented by ulcerative colitis and Crohn’s disease; and colorectal cancer (CRC). Dysbiosis can disrupt the mucosal barrier, resulting in perpetuation of inflammation and carcinogenesis.The increase in some specific groups of harmful bacteria, such as Escherichia coli (E. coli) and enterotoxigenic Bacteroides fragilis (ETBF), has been associated with chronic tissue inflammation and the release of pro-inflammatory and carcinogenic mediators, increasing the chance of developing CRC, following the inflammationdysplasia-cancer sequence in IBD patients. Therefore, the aim of the present review was to analyze the correlation between changes in the gut microbiota and the development and maintenance of IBD, CRC, and IBD-associated CRC. Patients with IBD and CRC have shown reduced bacterial diversity and abundance compared to healthy individuals, with enrichment of Firmicute sand Bacteroidetes.Specific bacteria are also associated with the onset and progression of CRC, such as Fusobacterium nucleatum, E. coli, Enterococcus faecalis, Streptococcus gallolyticus,and ETBF. Future research can evaluate the advantages of modulating the gut microbiota as preventive measures in CRC high-risk patients, directly affecting the prognosis of the disease and the quality of life of patients.
Key Words: Gut microbiota; Dysbiosis; Ulcerative colitis; Crohn’s disease; Inflammatory bowel disease; Colorectal cancer
lNTRODUCTlON
The human microbiome is composed of several types of microbes that colonize different niches of the human body, such as the skin, lungs, vagina, and gastrointestinal tract[1]. Of these, the gut microbiota is the most studied, due to the presence of greater diversity and number of microbial species compared to other parts of the body[1,2]. The gut microbiota is a complex community of approximately 100 trillion microorganisms, such as fungi, viruses, and protists, that inhabit the digestive tracts of humans, living in symbiosis with the host[3,4].
Alterations in the gut microbiota are associated with several diseases, including inflammatory bowel disease (IBD) and colorectal cancer (CRC)[2]. Thus, studies aimed at elucidating the role of the microbiota in each phase of each disease are necessary. Based on changes in the intestinal microbiota,new diagnostic tools and possible treatments can be developed. Further, studies aiming to correlate alterations in the microbiota or in specific bacterial populations with the diagnosis, prognosis, and/or treatment of diseases are necessary. Therefore, this mini-review was conducted to determine the correlation between changes in the gut microbiota and IBD, CRC, and IBD-associated CRC.
At birth, the intestinal tract is sterile[2], with bacterial colonization beginning as early as during the passage of the baby through the birth canal[5]. Colonization continues through feeding and other environmental contacts. Several factors are known to influence colonization, including gestational age,mode of delivery (vaginal birthvscesarean delivery), diet (breast milkvsformula) and exposure to antibiotics, and sanitation[2].
During the first year of life, the microbial composition of the mammalian intestine is relatively simple and characterized by low diversity with relative dominance of species from the phylaProteobacteriaandActinobacteria[2,5]. Over time, the microbiota becomes more diverse, with theFirmicutesandBacteroidetesphyla later emerging as the predominant microbes characterizing the adult microbiota[2,6-8].
In an adult individual, over 90% of intestinal bacteria belong toFirmicutes,Bacteroidetes,Proteobacteria,orActinobacteriaphyla[4,7]. The gut microbiota can be differentiated into beneficial bacteria and opportunistic bacteria, the latter of which can cause infection[8].Lactobacillus,Bifidobacterium,Enterococci,andPropionobacteriaare some of the beneficial microbes, while the opportunistic groups includeBacteriodes,Bacilli,Clostridia,Enterobacteria,Actinobacteria,Peptococci,Staphylococci, andStreptococci[8].
The benefits of the gut microbiota to the host’s physiology are vast, including nutrition, immune development, and host defense[4,7]. In the field of nutrition, some bacterial species, such asBifidobacterium, participate in the biosynthesis of several components as vitamin K and vitamin B[7]. In addition,some can provide short-chain fatty acids (SCFAs) by fermenting dietary fibers[7,9]. SCFAs are a group of small fatty acids (2 to 5 carbons) produced by anaerobic microorganisms that, after absorption, have systemic immunomodulatory and anti-inflammatory properties[9].
The most abundant SCFAs in the colon are acetate, propionate, and butyrate[7,9,10], which promote the proliferation of beneficial bacteria and stimulate regulatory T cells to reduce inflammatory mediators, associated with an increase in colonic oxygen consumption by epithelial cells, and enhancing immune regulation, and gut barrier function[9-11].
Imbalance between the beneficial and opportunistic groups causes dysbiosis, which is associated with gastrointestinal disorders such as IBS, IBD, and CRC[4,7,8].
lBD AND GUT MlCROBlOTA
IBD, a heterogeneous chronic and relapsing inflammatory illnesses of the digestive system, is traditionally classified into ulcerative colitis (UC) and Crohn’s disease (CD)[4,12,13]. Both UC and CD can cause several symptoms like diarrhea, rectal bleeding, and abdominal pain. However, inflammation in CD is transmural and can involve any part of the gastrointestinal tract. In contrast, inflammation in UC is more superficial and limited to the colon[14].
Although the cause of IBD is not fully known, genetic history is likely involved in the pathophysiology of IBD, thus, several studies have identified a number of susceptible genes[12]. However,environmental factors, such as stress, sleeping patterns, antibiotic use, hygiene, diet, and smoking, are also associated with the development of IBD[4].
While IBD is a disease with the highest rates in industrialized countries, its incidence varies considerably worldwide, and newly industrialized areas such as India and South America presented a rapid increase in case numbers[7,13,14]. This enhancement can be related to dietary habits such as increased consumption of processed foods, sugars, and fats; overutilization of antibiotics; and an overall improvement in sanitary conditions and hygiene[13].
Nevertheless, the exact factors that trigger the first episode of inflammation and subsequent relapse remain unknown, it is now know that inflammatory episodes can result, in genetically predisposed people, from an immune response against intestinal microbial antigens under several environmental conditions[14]. Several IBD-associated susceptible genes are correlated with host responses to gut bacteria, suggesting that the gut microbiota also participate in the pathogenesis of IBD[7]. Dysbiosis is a reduction in microbial diversity, or a combination of the loss of beneficial bacteria and an increase in pathogenic bacteria[15], that may influence the pathogenesis of IBD.
The most prominent changes in the microbiota of IBD patients are the decreased diversity in bacteria species associated with decreased abundance ofBacteroidetesandFirmicutes[4,7] alongside an increase in the abundance ofProteobacteria[4].
In patients with IBD, the mucus layer is compromised, allowing luminal bacteria to penetrate and invade the submucosal layers, leading to the proliferative and inflammatory processes[4,13]. Then,mucosal destruction due to inflammatory injury further exposes the submucosa to more bacteria,leading to a vicious, positive feedback cycle of antigenic exposure and mucosal damage[13].
Faecalibacterium prausnitziiis one of the most abundant human gut bacteria and can be used as synonym of the gut health because of its anti-inflammatory and immunoregulatory properties[14]. One of the its products is butyrate, which is associated with an anti-inflammatory effect[7]. Several authors have correlated decreases inFaecalibacterium prausnitzii(F. prausnitzii) abundance with CD development[7,14,16] and in UC patients, a decrease was also observed during remission, being the recovery of theF.prausnitziipopulation associated with the maintenance of clinical remission[17].
Roseburiaspp, includingRoseburia faecis,Roseburia intestinalis(R. intestinalis),and Roseburia hominis(R.hominis), another genus of butyrate-producing bacteria, were significantly lower in healthy individuals with a high genetic risk for IBD compared to healthy individuals with low genetic risk[7].R. hominis,specifically, was significantly reduced in UC patients, and an inverse correlation was observed betweenR hominisand disease severity[18]. In contrast,R. intestinalissupernatant suppressed the expression of interleukin (IL)-6 as well as the signal transducer and activator of transcription 3 (STAT3)viamacrophage regulation in anin vitroexperiment. Additionally, in dextran sodium sulfate- and 2,4,6-trinitrobenzene sulfonic acid-induced intestinal inflammation models,R. intestinalissupernatant reduced macrophages and Th17 cells in the colon, which was associated with the downregulation of IL-6 and STAT3[19].
In the field of pathogenic bacteria, several authors have described a relative increase inProteobacteria,mainlyEscherichia coli(E. coli), in IBD patients[7,20-22]. The exact mechanisms that lead to an increase inProteobacteriaduring inflammation are not completely understood; however, Rizzattiet al[20] proposed two mechanisms: The oxygen hypothesis and the presence of nitrate. In a normal colon, epithelial cells deplete oxygen in the lumen through beta oxidation, creating an anaerobic environment[23]. In contrast,during an inflammatory episode, the beta oxidation capacity of colonic cells is decreased, thus increasing oxygen availability, which promotes dysbiosis andProteobacteriagrowth[24]. Nitrate generated as a by-product during the inflammatory process conferred a growth advantage to the commensal bacteriaE. coliin the large intestine, which then became predominant[25].
The increase in pathogenic bacteria in the digestive tract that can adhere to the colonic mucosa could alters the gut permeability, modifying the diversity and composition of intestinal microbiota, and ultimately leading to intestinal inflammation by regulating inflammatory genes expression[7].
Dysbiosis can further alter bacterial metabolites; decreased concentration of SCFAs has been reported as a result of a diminish in butyrate-producing bacteria likeF. prausnitzii, in patients with IBD and in animal models of intestinal inflammation[7,9,10]. Decreased levels of SCFAs affects the differentiation and expansion of Treg cells and the growth of epithelial cells[26], leading to the loss of intestinal homeostasis.
CRC AND GUT MlCROBlOTA
CRC is the third most common cancer, and the second most frequent cause of cancer deaths worldwide being more common in developed than in developing countries[27,28]. Notwithstanding cases in all countries are on the rise, most CRC cases occur in Western countries, where there is an increase in incidence[28]. CRC rates in older adults in the United States have declined in recent years probably due to increased screening, but rates in younger adults have been rising, which may be correlated with higher incidence of obesity and other diet and lifestyle trends in the western hemisphere[29].
The exact mechanism for CRC onset and progression has not been fully elucidated, but it is generally believed to be the result of extensive and complex interactions between genetic and environmental factors[30]. According to Olovoet al[28], the progression of adenoma to carcinoma could be linked to the gut microbiota and conversely, a healthy microbiota is correlated with a minor risk of advanced adenoma[28].
Several factors that affect the gut microbiota are thought to be related to colon carcinogenesis, such as obesity, a high-fat diet, smoking, and frequent consumption of alcohol[30].
Patients with CRC have shown reduced bacterial diversity and abundance compared to healthy individuals, with enrichment ofFirmicutesandBacteroidetes[31]. Specific bacteria are also associated with the onset and progression of CRC, such asFusobacterium nucleatum,E. coli,Enterococcus faecalis,Streptococcus gallolyticus, and enterotoxigenicBacteroides fragilis(ETBF)[32].
The abundance ofFusobacterium nucleatum, an opportunistic pathogen found normally in the oral cavity, is correlated with age and tumor diameter in CRC patients[31,33,34]. Additionally, an overabundance ofF. nucleatumis associated with poor prognosis in metastatic CRC[32]; thus,F. nucleatumcould be considered a potential biomarker for predicting the prognosis in patients with proximal colon cancer[34].
There is plenty of evidence of the tumor-promoting effects ofStreptococcus gallolyticusin colon cells[35-37]. Colonic cells incubated withStreptococcus gallolyticus(S. gallolyticus) presented increased levels of β-catenin, c-Myc, and proliferating cell nuclear antigen (PCNA), all of which are transcription factors associated with cancer development[35]. In addition, in mice, administration ofS. gallolyticusleads to more tumors, higher tumor burden, and dysplasia grade, and increased cell proliferation and β-catenin staining in colonic crypts compared to mice incubated with control bacteria[35]. Furthermore, CRC patients present high levels of this bacterium compared to healthy individuals[37]. However, CRCspecific conditions, such as the increased concentration of bile acids, could also promoteS. gallolyticuscolonization, creating a maintenance cycle for high levels of this bacterium in the gut[37].
ETBF is the most frequent anaerobe isolated from cases of diarrhea, peritonitis, intra-abdominal abscesses, and sepsis, and there is a positive correlation between the presence of ETBF and active IBD and CRC[38]. According to Appunniet al[39], co-colonization of toxigenicE. coliand ETBF in mice resulted in increased production of pro-inflammatory IL-17 and subsequent DNA damage, which could accelerate the development of CRC[39]. In addition, the toxin in ETBF could induce c-myc expression and IL-8 secretion causing oxidative DNA and epithelial barrier damage, and STAT3/Th17 immune responses activation, which are further correlated with an increased risk of CRC[38].
lBD-ASSOClATED CRC AND GUT MlCROBlOTA
Intestinal inflammation is one of the most common risk factors for developing CRC. Besides being two to six times more likely to develop CRC than healthy people, IBD patients with cancer are affected younger than sporadic CRC patients[40]. Disease duration, extension of the lesions, inflammation,sclerosing cholangitis, age at onset and onset in childhood are factors linking IBD with an increased incidence of CRC[41,42].
Chronic inflammation initiates and drives tumorigenesis[42], leading to an “inflammation-dysplasiacarcinoma” sequence, not the “adenoma-sequence” classically described in sporadic CRC[40].
Popovet al[43]proposed the three main theories about bacterial involvement in the development of IBD-associated CRC: The alpha-bug hypothesis, driver-passenger hypothesis, and common ground hypothesis. In the alpha-bug hypothesis, a single bacterium (normally ETBF) is thought to cause all modifications and damage that lead to carcinogenesis. The driver-passenger hypothesis is similar, but after the attack by the first bacteria, other opportunistic bacteria start to grow and contribute to cancer development. Finally, in the common ground hypothesis, endogenous and exogenous factors form a“leaky gut,” allowing the passage of bacteria into the submucosal tissue, leading to chronic inflammation and the consequent emergence of cancer[43].
It is known that bacteria, through pathogen-associated molecular patterns (PAMPs), are capable of communicating with the tool-like receptors (TLRs), retinoic acid-inducible gene I-like receptors (RLRs),and nucleotide binding oligomerization domain-receptors (NLRs), and trigger an immune response[42]. Nuclear factor kB (NF-kB) can be activated by the TLR and tumor necrosis factor-α (TNF-α),inducing the transcription of several tumorigenesis genes such as COX-2, which then leads to the apoptosis of intestinal epithelial cellsviatumor suppressor p53 pathways[40] and consequent breakdown of the intestinal barrier, allowing microbial translocation.
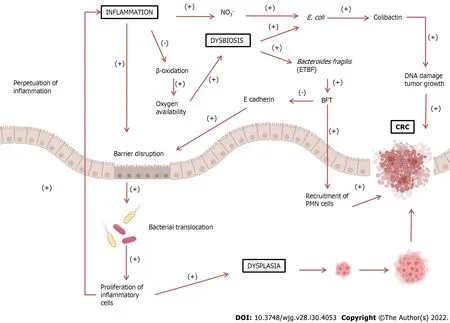
Figure 1 lncreased inflammatory response can lead to barrier disruption, allowing bacterial translocation into the intestinal lumen. These pathogens trigger an immune and inflammatory response, perpetuating the inflammatory process. Inflammatory cells can also cause dysplasia, leading to the development of colorectal cancer (CRC). Under normal conditions, epithelial cells, during β-oxidation, decrease oxygen availability, thus creating an anaerobic environment. During chronic inflammation, β-oxidation decreases, enhancing oxygen availability. This also leads to an increase in nitrate (NO3-) formation, leading to dysbiosis and growth of proteobacteria such as Escherichia coli (E. coli). E. coli produces colibactin, which could damage DNA and stimulate tumor growth. The growth of Bacteroidetes fragilis may also occur, which produces Bacteroides fragilis toxin (BFT). This toxin cleaves E-cadherin, a major constituent of the zonula adherens, which is responsible for cell adhesion, leading to further barrier disruption. Additionally, BFT also stimulates epithelial cells to recruit polymorphonuclear leukocytes (PMN) cells, promoting the development of CRC. BFT: Bacteroides fragilis toxin; ETBF: Enterotoxigenic Bacteroides fragilis; CRC: Colorectal cancer;PMN: Polymorphonuclear leukocytes.
Increased quantities ofProteobacteriaare associated with decreased levels of SCFAs and increased colonic inflammation[43]. In addition, intestinal Dysbiosis and barrier defects, associated with alterations in mucin secretion may happen even in the absence of active inflammation[43], perpetuating a favorable environment for the emergence of CRC.
Several studies have demonstrated the participation of bacteria such asE. coli,ETBF,andFusobacterium nucleatumin chronic inflammation and cancer development in IBD patients[40,42-45].
A review by Yuet al[44] concluded that commensal bacteria,E. coliandE. faecalis,isolated from healthy subjects are capable of initiating intestinal inflammation in genetically deficient mice,suggesting that opportunistic commensal bacteria may become pathogenic only in genetically predisposed hosts[44]. Nevertheless, some types ofE. colican produce colibactin, a genotoxic compound capable of promoting tumor growth, alkylating DNA, and inducing tumorigenic double-stranded breaks[45]. According to Yanget al[45], inflammation may upregulate bacterial virulence genes, such as colibactin, and facilitate colonization of the mucosa, leading to increased colibactin-induced DNA damage in colonic epithelial cells, allowing this bacterial strain to exert its carcinogenic activity[42].
The enterotoxin produced by ETBF known as bacteroides fragilis toxin, acted as a metalloprotease cleaving E-cadherin, a major constituent of the zonula adherens, which is responsible for cell adhesion[46]. This toxin can trigger an inflammatory, pro-tumoral signaling caspase in colonic epithelial cells that cause the recruitment of polymorphonuclear immature myeloid cells to promote colon cancers[44].
CONCLUSlON
There is a consensus that dysbiosis is present in both IBD and CRC, and that dysbiosis could lead to a disruption of the mucosal barrier with perpetuation of inflammation and carcinogenesis. Dysbiosis with a consequent increase in bacteria, such asE. coliand ETBF, is believed to lead to a breakdown of the intestinal mucosal barrier, allowing the translocation of more bacteria from the lumen to the interior of the tissue. This condition leads to chronic tissue inflammation, with the release of inflammatory and pro-carcinogenic mediators increasing the risk of developing CRC. This positive feedback loop of dysbiosis could be the basis for the inflammation-dysplasia-cancer sequence (Figure 1).
Further studies should be carried out to identify which bacteria or which set of bacteria may be responsible for this feedback cycle. Understanding the mechanism behind dysbiosis cycles may be the basis for preventing or treating IBD-associated CRC and CRC. Future research should evaluate the advantages of modulating the intestinal microbiota as a protective factor for the development of IBDassociated CRC; thus, the use of prebiotics, probiotics, or diet-based treatment can be used as preventive measures in CRC high-risk patients, directly affecting the prognosis of the disease and the quality of life of patients living with IBD.
FOOTNOTES
Author contributions:Quaglio AEV, Grillo TG, and De Oliveira ECS performed the majority of the writing; Di Stasi LC and Sassaki LY designed the outline and coordinated the writing of the paper; all authors critically revised the manuscript for important intellectual content and approved the final version.
Conflict-of-interest statement:There are no conflicts of interest to report.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Brazil
ORClD number:Ana Elisa Valencise Quaglio 0000-0002-5998-2382; Thais Gagno Grillo 0000-0002-4351-5034; Ellen Cristina Souza De Oliveira 0000-0001-5357-3468; Luiz Claudio Di Stasi 0000-0002-7864-1073; Ligia Yukie Sassaki 0000-0002-7319-8906.
S-Editor:Chen YL
L-Editor:A
P-Editor:Chen YL
 World Journal of Gastroenterology2022年30期
World Journal of Gastroenterology2022年30期
- World Journal of Gastroenterology的其它文章
- Role of one-step nucleic acid amplification in colorectal cancer lymph node metastases detection
- Current perspectives on the role of liver transplantation for Langerhans cell histiocytosis: A narrative review
- Thrombocytopenia in chronic liver disease: Physiopathology and new therapeutic strategies before invasive procedures
- P2X7 receptor blockade decreases inflammation, apoptosis, and enteric neuron loss during Clostridioides difficile toxin A-induced ileitis in mice
- Serological profiling of Crohn’s disease and ulcerative colitis patients reveals anti-microbial antibody signatures
- Trends in medication use and treatment patterns in Chinese patients with inflammatory bowel disease