
多步旋涂过程中CsPbBr3 无机钙钛矿成膜机理*
2022-08-12 14:28马书鹏林飞宇罗媛朱刘郭学益杨英
物理学报 2022年15期
马书鹏 林飞宇 罗媛 朱刘 郭学益 杨英†
1) (中南大学冶金与环境学院,长沙 410083)
2) (中南大学,有色金属资源循环利用湖南省重点实验室,长沙 410083)
3) (有色金属资源循环利用湖南省工程研究中心,长沙 410083)
4) (广东省高性能薄膜太阳能材料企业重点实验室,清远 511517)
5) (清远先导材料有限公司,清远 511517)
在无机钙钛矿太阳能电池的研究中,薄膜制备工艺是影响钙钛矿太阳能电池光电转换效率(PCE)的重要因素之一.CsPbBr3 钙钛矿作为稳定性极好的无机钙钛矿之一,因其前驱体盐(PbBr2,CsBr)溶解度差异过大,通常采用多步法进行制备.而由于对成膜机理的认识不充分,导致制备的薄膜存在薄膜形貌差、前驱体反应不完全等问题.本文通过旋涂不同次数的CsBr 溶液,探究了CsPbBr3 钙钛矿的成膜机理.成膜过程中CsBr 扩散进入预先沉积的PbBr2 薄膜完成反应,短暂反应时间使薄膜深层反应不充分而薄膜表面过度反应,CsPb2Br5 和Cs4PbBr6 等相伴随CsPbBr3 钙钛矿出现,反复退火形成的薄膜阻挡CsBr 扩散加剧了这一现象.适当地延长前驱体的反应时间,能为CsBr 扩散及反应提供更充分的空间.基于优化反应时间,CsPbBr3 钙钛矿薄膜形貌得到改善、其晶粒尺寸得到提升,钙钛矿薄膜中的晶界减少,从而抑制了载流子复合.在4 次旋涂和30 s 反应时间的条件下,组装的CsPbBr3 钙钛矿太阳能电池开路电压从1.01 V提升至1.28 V,PCE 从5.32%提升至6.30%,器件短路电流密度Jsc=8.40 mA/cm2,填充因子FF=59%.基于以上研究,为多步旋涂法制备CsPbBr3 钙钛矿薄膜和电池提供了理论借鉴.
1 引言
近年来钙钛矿太阳能电池引起了极大的研究热度,其具有载流子迁移率高、载流子长扩散、带隙小、吸光系数高等特点,光电转换效率(PCE)从3.8%提升至25.5%[1,2].水、氧气导致的电池衰退限制了钙钛矿的应用,因此大量研究工作集中在改善有机无机杂化钙钛矿太阳能电池稳定性的研究方面[3−7].Yang 等[8]通过引入琼脂糖,未封装器件能在空气湿度条件下维持90%以上效率超过1392 h.Gu-SCN (硫氰酸胍,guanidinium thiocyanate)作为添加剂也用于稳定MAPbI3(CH3NH3PbI3),SCN–与CH3NH3+反应在晶界处产生PbI2将提升钙钛矿薄膜的结晶、晶粒大小及稳定性[9].另一方面,通过Cs+替换有机官能团后形成的无机钙钛矿稳定性得到改善[10,11].Bai 等[12]制备的基于CsPbI2Br电池PCE 可达14.81%,器件在25%—35%湿度的空气气氛中维持效率不衰减.Lin 等[13]对CsPb I2Br 钙钛矿开展了研究,在无手套箱的条件下制备了可稳定存在144 h 的CsPbI2Br 钙钛矿.
相比于CsPbI3和CsPbIxBr3-x,CsPbBr3钙钛矿具有更大的带隙(2.3 eV),大的带隙能够为电池提供超高的开路电压(理论值1.98 V),为CsPbBr3钙钛矿在光电方面的应用带来了巨大的优势.Cs PbBr3钙钛矿薄膜质量是影响电池PCE 的重要因素之一,探究其成膜机理将为制备高质量薄膜提供便利.由于CsBr 和PbBr2溶解度差异大,Kulbak等[14]提出分步法制备CsPbBr3钙钛矿薄膜,简单的旋涂薄膜中存在许多缺陷,电池PCE 仅5.98%.分步法过程中两前驱体盐浓度差会影响钙钛矿形成,CsBr 不足时薄膜中存在CsPb2Br5,过多的CsBr 又将导致薄膜中形成Cs4PbBr6[15,16].此外,CsBr 与PbBr2反应速度极快,沉积到基底上将导致部分PbBr2未完全反应,退火后形成的钙钛矿薄膜粗糙[17].限制CsBr 的沉积速度,为PbBr2提供足够的时间转变成CsPbBr3钙钛矿,才能够制备出表面平滑且粒径均匀的钙钛矿薄膜.目前CsPbBr3钙钛矿薄膜的研究多集中在工艺方法的优化上,旋涂、浸泡、气相沉积、脉冲激光沉积[18−20]都可用于薄膜制备,但对于CsPbBr3的形成机理研究仍然较少.
本文以旋涂法为基础,将CsBr 溶液反复旋涂在PbBr2薄膜上,观察其变化规律以探究CsPbBr3钙钛矿的形成机理.研究发现,钙钛矿薄膜中的相变过程为PbBr2→CsPb2Br5→CsPbBr3→Cs4PbBr6.由于每次旋涂后都进行退火,已形成的钙钛矿薄膜将阻碍底层未反应的PbBr2和CsPb2Br5转变为CsPbBr3钙钛矿.而CsBr 的溶剂甲醇对钙钛矿膜有刻蚀作用,可消除钙钛矿膜的阻碍.适当地延长CsBr 溶液在PbBr 薄膜表面的反应时间,可以溶解阻挡层,使下层PbBr2充分反应.同时,低浓度的CsBr 溶液不会导致Cs4PbBr6形成.通过对CsPbBr3薄膜的形成机理的深入探究及优化,在旋涂4 次及溶液停留30 s 的条件下,制备了致密且粒径均匀的钙钛矿薄膜,制备的结构为FTO/c-TiO2/m-TiO2/CsPbBr3/Spiro-OMeTAD/Ag 的电池PCE 可达6.30%.
2 实验部分
2.1 实验材料
刻蚀的FTO 导电玻璃购自大连七色光太阳能科技公司.钛酸四异丙酯(>99.5%),18NR-T(TiO2浆料),PbBr2(>99.99%),Sprio-OMeTAD(98%),TBP(4-tert-butylpyridine,AR),LITFSI((三氟甲基磺酰)亚胺锂),PC61BM(苯基c61 -丁酸甲酯,AR)购自西安宝莱特光电科技公司.CsBr(>99%),DMF(99%),氯苯(99%),乙腈(99%)购自百灵威试剂公司,甲醇(AR),乙醇(AR),正丁醇(AR)购自阿拉丁试剂.
2.2 电池制备
首先FTO 玻璃经过清洗剂、去离子水、乙醇超声清洗三次,在烘箱中80℃烘干.随后摩尔浓度为0.22 mol/mL 的钛酸四异丙酯溶液和质量比1∶4 的18NR-T 溶液(18NR-T,乙醇)分别旋涂到FTO 基底上制备电子传输层,旋涂速度分别为2000 和4000 r/min,旋涂时间为30 s,旋涂后分别经过500 ℃烧结使其形成TiO2.随后通过多步旋涂法制备钙钛矿薄膜:将摩尔浓度为1 mol/mL的PbBr2溶液以旋涂速度2000 r/min、旋涂时间30 s 的条件旋涂到TiO2电子传输层上制备PbBr2薄膜,在70 ℃加热30 min 烘干溶剂,然后将质量浓度为15 mg/mL 的CsBr 以2000 r/min 的转速旋涂30 s 与PbBr2薄膜反应,250 ℃退火5 min,随后多次重复这一过程制备CsPbBr3钙钛矿薄膜.对于空穴传输 层,质量浓度为72.3 mg/mL 的Spiro-OMeTAD(溶剂氯苯,添 加TBP,LITFSI(520 mg/mL,溶剂乙腈))以旋涂速度3000 r/min、旋涂时间30 s 的条件旋涂到钙钛矿薄膜表面.最后通过蒸镀仪(PD400s,普迪真空,中国)制备Ag 金属电极,即完成电池器件的制备.
2.3 表 征
钙钛矿吸光层的晶相结构和表面形貌采用X 射线衍射仪(日本,Rigaku-TTRⅢ)和场发射扫描电子显微镜(日本电子公司,JSM-6360LV)表征.对可见光吸收效率采用紫外-可见光分光光度计(UV-1800,Hitachi,日本)测试,扫描范围为400—1100 nm.使用具有466 nm 脉冲的FLS1000测量光致发光光谱(爱丁堡仪器,英国).采用电化学工作站(PGSTAT302N,Metrom,AUT86802,瑞士)和氙灯模拟光源(CHF-XM500,Trust-tech,北京)组合分析钙钛矿太阳能电池的光电性能(AM1.5G,100 mW/cm2).电化学阻抗(EIS)通过电化学工作站在暗态环境下测量,偏压设置为1 V.缺陷态密度采用仅有电子传输层的结构FTO/TiO2/CsPBbr3/PC61BM/Ag 通过空间电荷受限电流(SCLC)法在暗态环境下测试.电池器件有效面积0.1657 cm2.所有测试均在湿度低于40%的空气中进行.
3 结果与讨论
3.1 旋涂次数对钙钛矿薄膜的影响
图1(a)为多步旋涂法制备的CsPbBr3钙钛矿薄膜示意图,钙钛矿薄膜通过两步法进行制备.首先沉积PbBr2到表面覆盖TiO2的FTO 玻璃基底上,通过多次旋涂,PbBr2薄膜会逐渐转变为黄色,这意味着钙钛矿在不断形成,结果如图1(b)所示.
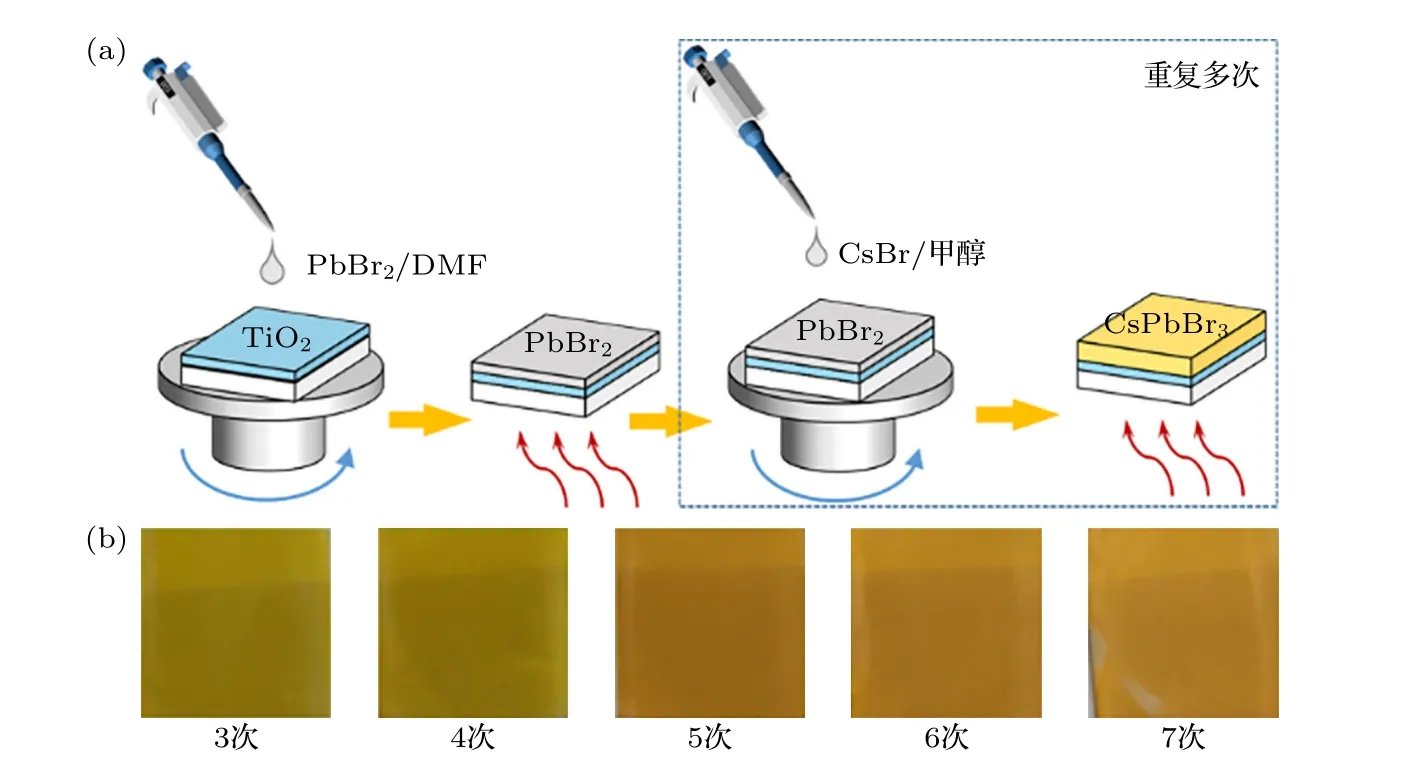
图1 (a) 多步旋涂法示意图;(b) 不同旋涂次数钙钛矿薄膜照片Fig.1.(a) Schematic of multi-step spinning method;(b) photographs of perovskite films with different times of spin coating.
对不同旋涂次数制备的钙钛矿薄膜进行X 射线衍射(XRD)测试,结果如图2(a)所示.在3—7 次旋涂次数下,薄膜都有位于15.40°,21.83°,30.90°,34.71°和44.32°的峰,分别对应CsPbBr3钙钛矿的(100),(110),(200),(210)和(220)晶面(PDF#54-0752).旋涂次数较少时,峰强度较低.所有薄膜均存在位于11.83°,23.61°,29.59°,40.81°的特征峰,分别对应CsPb2Br5的(002),(210),(213),(310)晶面(PDF#25-0211).结合其他研究,在溶液法制备CsPbBr3的过程中,CsPb2Br5总是存在[15,16,21].随着CsBr 不断地旋涂到薄膜表面,CsPb2Br5向CsPbBr3转化,XRD 结果中CsPbBr3位于15.40°,21.83°,30.90°的特征峰增强.但仅增加旋涂次数难以消除CsPb2Br5相,即使Cs4PbBr6相出现,Cs Pb2Br5仍然存在.
图2(b)和图2(c)为不同旋涂次数钙钛矿薄膜的紫外吸收光谱及相应的Tauc 图.在紫外光区域,所有旋涂次数制备的薄膜都具有较高的吸收值,其中旋涂4—5 次的薄膜吸收值要高于其他旋涂次数.4—5 次旋涂使PbBr2与CsBr 反应更充分,钙钛矿薄膜结晶更好.不同旋涂次数的薄膜吸收峰都位于535 nm,且没有发生偏移.通过计算可以发现,所有薄膜的带隙都处于2.33—2.34 eV 之间.图2(d)为不同旋涂次数下钙钛矿薄膜的光致发光(PL)光谱,薄膜在TiO2表面制备,具有电子传输层后,载流子可以进行分离.PL 光谱有两个明显的发射峰,其中左侧440 nm 为PbBr2/TiO2薄膜的发射峰(与图2(d)插图PbBr2的发射峰位置相同),而530 nm 发射峰为CsPbBr3钙钛矿发射峰[16].可以发现,旋涂4 次的钙钛矿薄膜具有最低的发射峰强度,这表明钙钛矿具有更好的电荷分离效果.
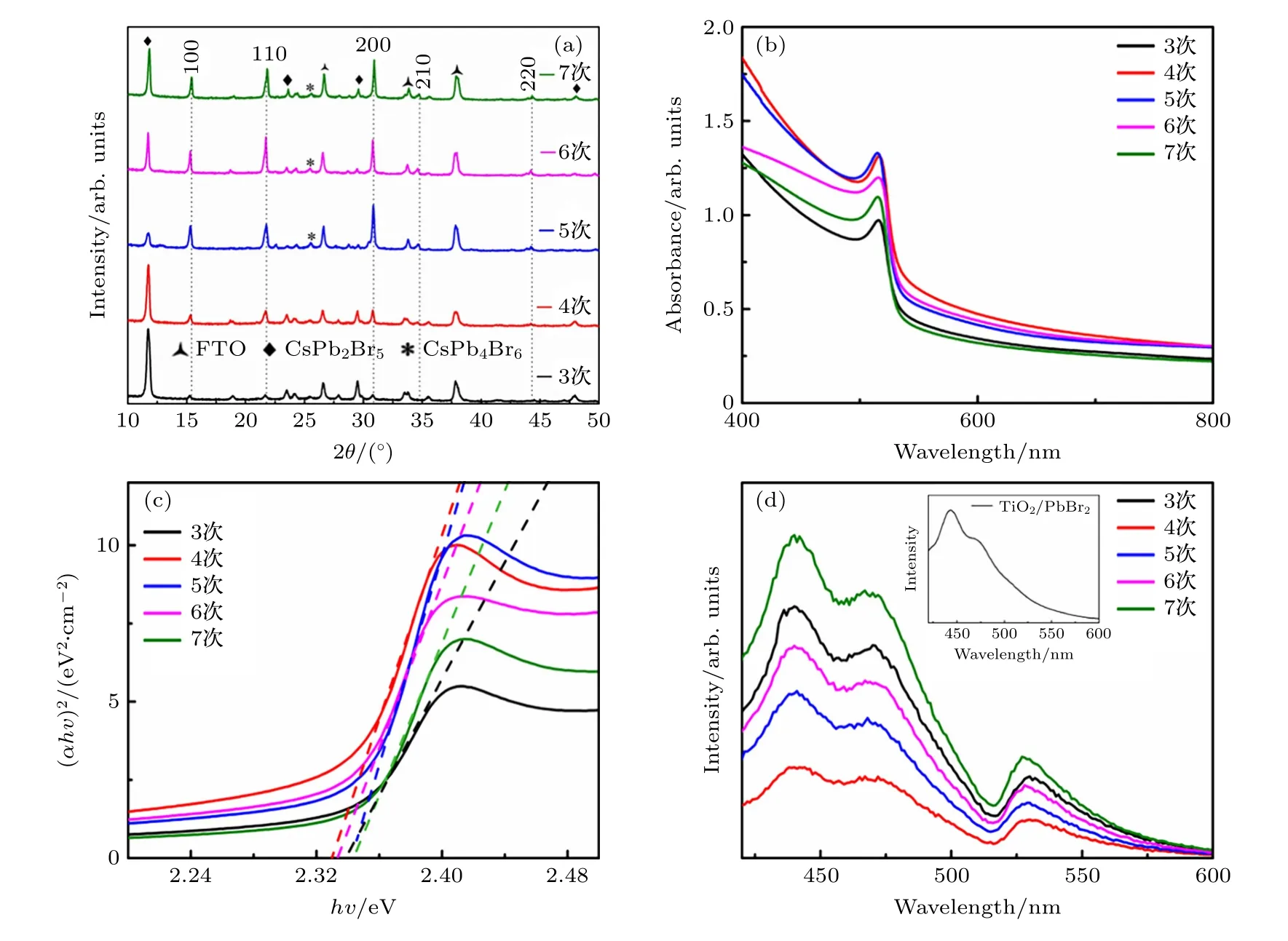
图2 不同旋涂次数CsPbBr3 钙钛矿薄膜的(a) XRD 图、(b) 紫外吸收光谱图、(c) Tauc 图和 (d) PL 光谱Fig.2.(a) XRD patterns,(b) UV-vis absorption spectra,(c) Tauc plots of (αhν)2 vs.the photo energy,and (d) PL spectra of CsPbBr3 perovskite films with different spin-coating times.
图3 为不同旋涂次数下钙钛矿薄膜的扫描电子显微镜(SEM)表面及截面图.不同旋涂次数下的薄膜形貌均较差,薄膜晶粒较小覆盖不完整且有孔洞.随着旋涂次数的增加,薄膜晶粒增大但仍不能完整覆盖表面.结晶差及过多的CsPb2Br5将导致晶粒较小[22].在薄膜未覆盖的区域,能够观察到一些未反应的PbBr2颗粒.图3(f)为旋涂4 次时薄膜的截面SEM 图,细小的颗粒堆积在一起印证了薄膜形貌和结晶差.同时说明钙钛矿薄膜表面和底层形貌存在巨大差异,这导致表面CsPbBr3完全形成,而底层的PbBr2未发生反应,CsPb2Br5始终存在于薄膜中.

图3 不同旋涂次数制备的CsPbBr3 钙钛矿薄膜SEM 图 (a) 旋涂3 次;(b) 旋涂4 次;(c) 旋涂5 次;(d) 旋涂6 次;(e) 旋涂7 次;(f) 旋涂4 次时薄膜的截面Fig.3.The SEM images CsPbBr3 perovskite films with different spin coating times:(a) 3 times;(b) 4 times;(c) 5 times;(d) 6 times;(e) 7 times;(f) cross-section image of the film with 4 times.
3.2 CsPbBr3 的成膜机理
结合薄膜照片,XRD,SEM 和其他研究[15,18,23],多步旋涂过程中CsPbBr3的形成机理可概况为以下3 个反应:
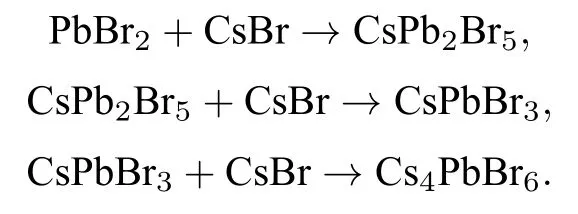
图4 展示了多步旋涂过程中CsPbBr3薄膜的形成机理.在预先沉积的PbBr2薄膜表面旋涂较少的CsBr 溶液时,CsPb2Br5形成占主导地位,已形成的CsPb2Br5将向下沉积.随着反复的旋涂CsBr 溶液,薄膜中发生相的融合分离,已形成的CsPb2Br5转化为CsPbBr3,同时底层未反应的PbBr2转变为CsPb2Br5.通过退火钙钛矿晶粒长大,形成紧密的钙钛矿薄膜覆盖在表面.当旋涂CsBr 过量后,Cs4PbBr6相出现.薄膜相变过程按照PbBr2→CsPb2Br5→CsPbBr3→Cs4PbBr6路线进行,这一结果在XRD 中得到了验证.但滴加CsBr 溶液后直接进行旋涂,已形成的钙钛矿薄膜将阻挡CsBr,使CsBr 难以进入到薄膜底层进行反应,这就导致薄膜中Cs4PbBr6出现时CsPb2Br5仍然存在.
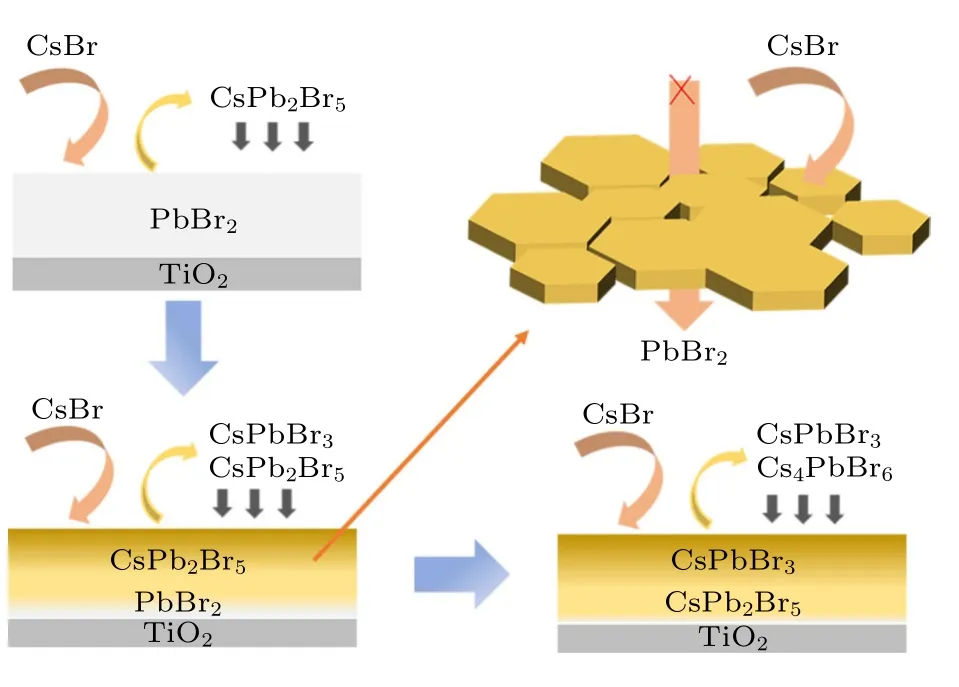
图4 CsPbBr3 钙钛矿薄膜的形成机理图Fig.4.Formation mechanism of CsPbBr3 in multi-step spin-coating.
3.3 CsBr 溶液反应时间对钙钛矿薄膜的影响
基于对旋涂次数因素的探索,薄膜在4 次旋涂时具有较好的形貌及光学性能.在此基础上探索CsBr 与PbBr2两前驱体反应时间对CsPbBr3钙钛矿薄膜形成及电池性能的影响.图5(a)为CsBr溶液被滴加到PbBr2薄膜表面后,两前驱体反应时间对薄膜颜色的影响.60 s 反应时间制备的样品黄色薄膜颜色最明亮,其他样品则随着反应时间减少不断变淡.为深入探究反应时间对薄膜的影响,对不同反应时间下退火与未退火的薄膜表面形貌进行SEM 测试,结果如图5(b)—(o)所示.图5(b)—(h)为未退火的薄膜SEM 图,薄膜覆盖度随反应时间的延长不断下降,这表明已形成的CsPbBr3薄膜被甲醇溶剂刻蚀加剧.甲醇对CsPbBr3的刻蚀作用在Ryu 等[24]的研究中也得到验证.图5(i)—(o)为退火后的薄膜,薄膜经过退火覆盖度明显上升.反应时间为0 时,薄膜覆盖度较好但晶粒较小,这是由于薄膜中存在大量CsPb2Br5.随着反应时间增加,钙钛矿晶粒明显增大,但当反应时间超过30 s 时,薄膜表面开始出现孔洞.延长反应时间能够加剧甲醇对已形成薄膜的刻蚀作用,使CsPb2Br5和PbBr2能够充分得到反应转化为CsPbBr3钙钛矿.

图5 (a) 不同反应时间下CsPbBr3 钙钛矿薄膜照片;(b)—(h) 未退火的薄膜SEM 图;(i)—(o) 退火后的薄膜SEM 图.标尺1 µm Fig.5.(a) Images of as-prepared films with varied CsBr solution reaction time;(b)–(h) SEM images of unannealed films;(i)–(o) SEM images of annealed films.All films spin-coating four times.Scale bar:1 µm.
对不同反应时间下形成的CsPbBr3钙钛矿薄膜进行XRD 测试,结果如图6(a)所示.在所有反应时间下,薄膜都存在位于15.18°,21.58°,30.69°,34.46°,44.29°的峰,分别对应CsPbBr3钙钛矿的(100),(110),(200),(210),(220)晶面.各个反应时间下,峰位没有发生偏移,同时这些峰的强度也随着反应时间的延长不断增强,60 s 时达到最大,表明延长反应时间有利于薄膜形成更多的CsPbBr3钙钛矿.但在所有反应时间下,位于11.7°,29.38°的峰一直存在,这两个峰分别对应CsPb2Br5相的(002),(213)晶面(PDF#25-0211).当反应时间到达60 s 时,CsPb2Br5峰强度最小,表明更长地反应时间能够有效地消除CsPb2Br5相.但由于薄膜表面和底层与CsBr 前驱体反应不完全均一,因此旋涂过程中CsPb2Br5难以完全消除,在3.1 节旋涂次数的实验中薄膜出现Cs4PbBr6时CsPb2Br5仍然存在,这和本节实验互相印证.同时少量的CsPb2Br5对于钙钛矿薄膜不完全有害,其掺在钙钛矿薄膜中能钝化CsPbBr3表面,降低CsPbBr3表面固有的Br 空位(VBr),提高载流子寿命[25].当反应时间超过40 s 时,在12.89°处出现了Cs4PbBr6的峰位[26],这表明反应时间大于30 s 后,CsBr 与PbBr2薄膜反应过度,导致CsPbBr3钙钛矿转变为其他相[15].
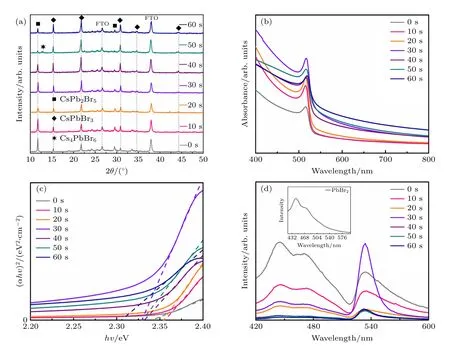
图6 不同反应时间下CsPbBr3 钙钛矿薄膜的(a) XRD 图、(b) 紫外吸收光谱、(c) Tauc 图和 (d) PL 光谱Fig.6.(a) XRD patterns,(b) UV-vis absorption spectra,(c) Tauc plots of (αhν)2 vs.the photo energy,and (d) steady-state PL of the cesium lead bromide films deposited on FTO substrates with varied CsBr solution reaction time.
图6(b)和图6(c)为薄膜的紫外吸收光谱和相应的Tauc 图,薄膜的吸收值随着反应时间的延长呈现先增强后减弱的趋势,在反应时间为30 s 时薄膜吸收值最大.同时所有反应时间下薄膜的吸收峰都位于535 nm,未发生偏移.30 s 具有较高吸收值说明PbBr2与CsBr 反应更充分,钙钛矿薄膜中其他相更少且结晶更好.通过带隙计算,所有反应时间下的样品带隙都在2.3 eV 左右.图6(d) 为PbBr2薄膜和不同反应时间钙钛矿薄膜的PL 光谱(样品直接沉积在玻璃基底上).反应时间小于30 s 时,PL 光谱中存在两个明显的发射峰,其中左侧440 nm 处的发射峰为PbBr2,右侧位于534 nm的峰则为CsPbBr3钙钛矿发射峰.反应时间从0 到60 s 过程中,左侧PbBr2发射峰强度不断下降,右侧钙钛矿发射峰强度不断上升.这说明延长反应时间能够有效地消除薄膜中的PbBr2促进钙钛矿形成.
基于以上研究制备CsPbBr3钙钛矿薄膜后,组装太阳能电池,结构为FTO/c-TiO2/m-TiO2/CsPbBr3/Spiro/Ag.在AM1.5G (100 mW/cm2)下测试的器件的J-V参数如表1 所列,包括Jsc,Voc,FF,PCE.图7 为不同反应时间下器件的J-V曲线,随着反应时间从0 增加到30 s,电池PCE 从5.32%增加到6.3%,此时Jsc=8.40 mA/cm2,Voc=1.28 V,FF=59%.进一步增加 反应时间到60 s,器件PCE 下降到2.09%.反应时间从0 延长到30 s,器件开路电压Voc提升至1.28 V.钙钛矿薄膜晶粒不断增大,从而有效地减少了晶界.钙钛矿薄膜的晶界会在其相关电荷陷阱态引起电荷复合,减少晶界将有利于器件获得更高的开路电压(Voc)和短路电流(Jsc)[26].同时,在30 s 反应时间下,钙钛矿薄膜具有最强的PL 发射峰,表明在这一条件下,钙钛矿结晶性能更好[17],从而为相应的器件提供了更高的性能参数.

表1 不同反应时间下CsPbBr3 钙钛矿薄膜的电池器件J-V 参数Table 1.J-V parameters of CsPbBr3 perovskite for solar cell with different reaction time.
EIS 测试用于探索延长反应时间前后电池的电荷传输过程,对未延长反应时间0 和最佳时间30 s 进行测试,结果如图7(b)所示.等效电路图如图7(b)插图所示,Rs和Rrec分别串联电阻和电荷复合电阻,数据也在图7(b)中给出.延长反应时间至30 s 后,Rs从27.39 Ω 降低至7.88 Ω,这表明电池的导电性能得到提升,对应着FF的提升.相比于反应时间为0,Rrec值从156.5 Ω 增大至1144 Ω,表明延长反应时间至30 s 载流子复合得到有效抑制,这有利于电荷的快速提取、分离,从而提高光伏性能[27,28].为进一步探索延长反应时间前后CsPbBr3薄膜的缺陷态密度,采用SCLC 测试记录的电池在暗态环境下的J-V曲线如图7(c)所示,器件结构为FTO/TiO2/CsPbBr3/PC61BM/Ag.缺陷态密度ntrap可由以下公式计算[29]:
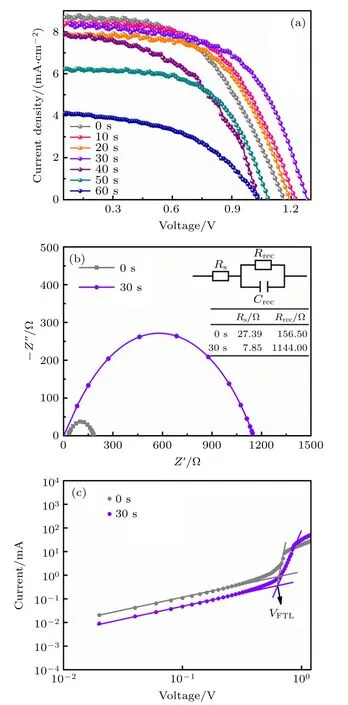
图7 (a) 不同反应时间下的CsPbBr3 钙钛矿薄膜器件J-V曲线;(b) CsPbBr3 电池Nyquist 图,插图为等效电路图及相关参数;(c) 暗态下结构为FTO/TiO2/CsPbBr3/PC61BM/Ag 的器件的J-V 曲线Fig.7.(a) J-V curves of CsPbBr3 perovskite solar cell based on different reaction time;(b) Nyquist plots of CsPbBr3 PSCs under 1 sun illumination,the inset provides the equivalent circuit and relevant parameter;(c) J-V curves of the device with an architecture of FTO/TiO2/CsPbBr3/PC61BM/Ag under dark conditions.
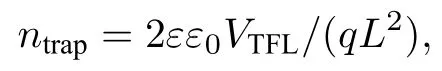
其中ε0是真空介电常数(8.85×10–12F/m),ε代表CsPbBr3的相对介电常数(≈ 22)[30],VTFL代表缺陷填充的限制电压,L为钙钛矿薄膜的厚度(约350 nm).在其他参数相同的条件下,VTFL越小钙钛矿薄膜的缺陷态密度越低.相比于未延长时间的CsPbBr3钙钛矿薄膜,反应时间延长至30 s 后,VTFL值从0.6494 V 下降0.6340 V,表明延长反应时间有利于减少钙钛矿薄膜中的缺陷态密度.通过一系列的探索分析,延长反应时间至30 s 是一种有效的提升钙钛矿薄膜质量,增强电池光电性能的有效手段.
4 结论
本文通过不同旋涂次数,探究了CsPbBr3钙钛矿薄膜的成膜机理,以及旋涂过程中CsBr 和PbBr2薄膜反应时间对钙钛矿薄膜和太阳能电池器件的影响.随着旋涂次数增加,钙钛矿薄膜的质量显著提升,旋涂过程中薄膜物相沿PbBr2→CsPb2Br5→CsPbBr3→Cs4PbBr6变化.已形成的钙钛矿薄膜会阻碍后续旋涂的CsBr 与PbBr2,CsPb2Br5充分反应,导致薄膜中同时存在CsPb2Br5和Cs4PbBr6.通过甲醇溶剂对CsPbBr3薄膜的刻蚀作用,延长CsBr 和PbBr2反应时间,能够使前驱体充分反应而不产生Cs4PbBr6.随着反应时间从0 延长到60 s,钙钛矿薄膜晶粒尺寸不断提升,但当反应时间过长时,致密的钙钛矿薄膜表面开始出现孔洞.综合以上因素,在30 s 反应时间下制备的电池器件具有最高的PCE.适当地延长反应时间后,CsBr 和PbBr2两种前驱体充分接触、反应,从而使其在薄膜中形成更多更好的形成CsPbBr3钙钛矿相,充分反应有利于减少CsPb2Br5相使薄膜形成更大的晶粒,减少晶界和缺陷态密度,抑制载流子的复合从而提升器件效率,经过优化最终制备了PCE=6.30%,Jsc=8.40 mA/cm2,Voc=1.28 V,FF=59%的基于CsPbBr3钙钛矿的太阳能电池器件.
猜你喜欢
无机化学学报(2022年8期)2022-08-09
电子测试(2022年12期)2022-07-18
无机材料学报(2022年1期)2022-04-12
燃烧科学与技术(2021年5期)2021-10-28
科学与财富(2021年33期)2021-05-10
考试与评价·高一版(2020年2期)2020-10-29
汽车零部件(2018年5期)2018-06-13
能源研究与信息(2014年3期)2014-10-30
建材发展导向(2014年2期)2014-05-04
电子世界(2004年5期)2004-07-26
