
基于Gaussian的甲烷爆炸微观反应计算分析*
2022-08-08 01:21王秋红蒋夏夏代爱萍
中国安全生产科学技术 2022年6期
王秋红,蒋夏夏,代爱萍
(1.西安科技大学 安全科学与工程学院,陕西 西安 710054;2.西安科技大学 化学与化工学院,陕西 西安 710054)
0 引言
瓦斯爆炸宏观特性及其微观反应机理一直是重点研究方向[1-5]。瓦斯的主要成分是甲烷(CH4),目前研究得出的几种CH4燃烧反应机理中[6-9],GRI-Mech 3.0机理[10]已经过大量实验验证,是目前国际公认的机理[11]。此后有学者对GRI-Mech 3.0机理进行了简化[12-14],为防治瓦斯爆炸事故提供了理论支撑。
近代量子化学计算的发展为机理探索提供了新的研究手段,He等[15]采用分子动力学模拟(MD)和密度泛函(DFT)理论计算了甲烷爆炸过程,发现CH3·和HO2·对爆炸反应速度有显著影响,OH·是爆炸过程中最重要的自由基。Luo等[16]利用Gaussian软件确定了CH4,CO,C2H6,C2H4,H2气体混合体系爆炸的热力学和动力学特性,研究得出5种可燃气体联合爆炸的起爆机理和一次起爆途径;以及磷酸二氢铵粉末抑爆甲烷爆炸的反应机理。罗振敏等[17-23]采用Gaussian软件定量分析了氨气(NH3)阻尼CH4爆炸的微观基元反应,及CO2/CH4,CO/CH4,C2H4/CH4混合体系爆炸微观机理,揭示了NH3,CO2,CO对CH4爆炸链式反应机理的阻尼作用,总结出C2H4/CH4混合爆炸的简化机理。姜海洋等[24]使用Gaussian软件分析了CO,H2O对甲烷爆炸链式反应的抑制作用,发现CO,H2O均会消耗CH4爆炸反应中的中间体及自由基。
以上研究在采用Gaussian软件分析惰性气体或可燃气体对CH4爆炸微观反应机理的影响时,忽略了体系中极化和弱相互作用对计算结果的影响。基于此,本文考虑体系中存在的弱相互作用对结构优化的影响,使用包含色散矫正的密度泛函理论DFT-D3(BJ)对侯金丽等[12]提出的甲烷爆炸反应简化机理中全部基元反应的反应物、中间体、过渡态、产物进行全参数构型优化,确定能量最低构型与过渡态的结构,并在相同水平下进行内敛反应坐标(IRC)计算,验证反应通道,从热力学及动力学角度对CH4爆炸微观反应简化机理进行分析,是对甲烷爆炸微观反应机理的更进一步深入研究。
1 建立模型
1.1 甲烷爆炸微观反应机理
侯金丽等[12]根据稳态燃烧温度及主要组分浓度分布,利用敏感性分析方法将GRI-Mech 3.0机理简化,如表1所示。
1.2 计算方法
以表1所述CH4燃烧反应简化机理为基础,应用量子化学计算软件Gaussian,考虑体系中存在的极化和弱相互作用,使用DFT-3色散矫正中的阻尼形式(BJ-

表1 CH4爆炸微观反应简化机理
damping),即DFT-D3(BJ),在B3LYP-D3(BJ)/6-31+G*水平下对甲烷爆炸微观反应机理中各反应物、中间体、过渡态、产物进行构型优化。由于理论计算忽视非谐振效应以及理论方法自身存在的误差会造成计算出的基频、零点能、焓、熵都可能有较大误差,对理论计算出的谐振频率乘以频率矫正因子,从而使得计算出来的基频及热力学校正量更加精准,用于获得准确焓变的频率校正因子为1.005 3,零点能校正因子为0.982 9,熵校正因子为1.008[25],频率计算稳定点为全实频,过渡态有且只有1个虚频。M06-2X泛函自身包括氢键的弱相互作用和色散矫正,使用M06-2X/def2-tzvpp计算电子能量E,通过以下计算得到反应的ΔH,ΔS,ΔG。理论计算如式(1)~(3)所示:
ΔH=H(T)-H(0)
(1)
H(0)=U(0)=E+ZPE
(2)
G=E+ZPE+ΔH-T·S
(3)
式中:H为焓,J;ΔH为焓变,kJ·mol-1;E为电子能量,kJ·mol-1;ZPE为真空零点能kJ·mol-1;G为吉布斯自由能,kJ·mol-1;T为温度,K;S为熵,kJ·K-1。
结构优化时自洽场的4个收敛条件必须同时满足:最大受力(Maximum Force)<0.000 450,均方根受力(RMS Force)<0.000 300,最大位移(Maximum Displacement)<0.001 800,均方根位移(RMS Displacement)<0.001 200。
2 结果与分析
2.1 甲烷爆炸微观反应历程分析
应用TS方法确定表1中CH4爆炸各基元反应的过渡态,可知:反应1为自由基结合反应,无过渡态;反应9为分解(分裂)反应,无过渡态;反应2,3,4,5,6,7,8,10分别对应过渡态TS(2),TS(3),TS(4),TS(5),TS(6),TS(7),TS(8),TS(10),且频率计算结果有且只有1个虚频,分别为-1 115.09,-137.33,-481.93,-919.55,-1 318.83,-1 921.16,-610.27,-1 491.89 icm-1,并通过内敛反应坐标(IRC)验证反应通道,即过渡态的两端分别连接着反应物和产物。
2.1.1 几何构型优化与化学键演变分析
通过构型优化得到表1甲烷微观爆炸反应机理中各驻点稳定构型及其参数,如图1~2所示。
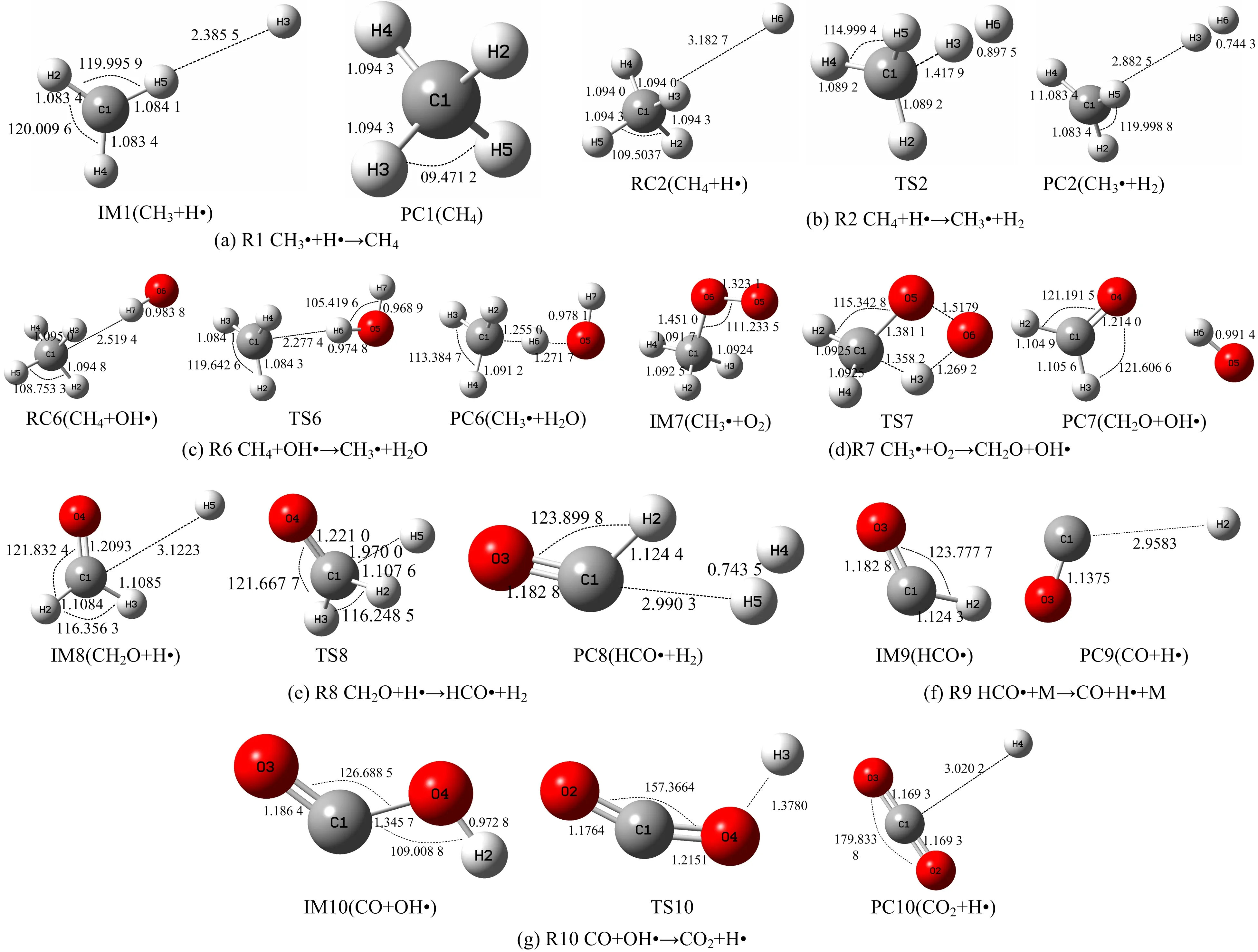
图1 甲烷氧化路径各驻点的稳定构型(键长/Å,键角/(°))
由图1(b)可知:反应2 CH4+H·→CH3·+H2中,CH4上的1个C-H键长逐渐增大脱H,同时R(H3,H6)距离不断减小,达到成键距离后生成H2和CH3·。由图1(c)可知,反应6 CH4+OH·→CH3·+H2O中,R(C1,H2)从1.094 8Å增加到1.255 0Å,C-H键长不断增大,作用力减弱,H从CH4上脱落,同时R(O5,H6)的距离不断减小,达到成键距离后,H与OH·上的O结合生成H2O。由图1(d)可知,反应7 CH3·+O2→CH2O+OH·中,R(C1,H3)从1.092 4Å,增加到1.358 2Å,C-H键长不断增大,作用力减弱,H从CH3·上脱落,同时R(O5,O6)从1.323 1Å增加到1.517 9Å后断裂,其中1个O与CH3·上脱掉的H结合生成OH·,同时R(C1,O5)的距离缩短,达到成键距离后生成CH2O。由图1(e)可知,反应8 CH2O+H·→HCO·+H2中,R(C1,H3)从1.108 5Å缩短到1.107 6Å,H从CH2O上脱落,同时R(H3,H5)键长不断缩短,达到成键距离后生成H2。由图1(g)可知,反应10 CO+OH·→CO2+H·中,R(O4,H2)从0.972 8Å增加到1.378 0Å,O-H键长不断增大,作用力逐渐减弱,使得H从OH·脱离,同时R(C1,O4)从1.345 7Å缩短到1.215 1Å,达到成键距离后O与CO上的C结合生成CO2。
由图2(a)可知,反应3 O2+H·→OH·+O·中,R(O1,O2)从1.333 0Å增加到2.244 2Å,O-O键长增大,作用力减弱,脱落1个O,同时R(O1,H3)逐渐缩短,达到成键距离后生成OH·。由图2(b)可知,反应4 O·+H2→OH·+H·中,R(H1,H2)从0.763 8Å增加到0.776 9Å,H-H键长不断增大,作用力减弱,脱落1个H与反应物中的O·结合生成OH·和H·。如图2(c)所示,反应5 OH·+H2→H2O+H·中,R(H1,H2)从0.743 7Å增加到0.837 1Å,H-H键长不断增大,作用力减弱,H从H2脱落,与OH·中的O结合生成H2O。

图2 反应3,4,5各驻点的稳定构型(键长/Å,键角/(°))
2.1.2 甲烷氧化路径分析
结合以上对甲烷氧化过程中键的演变分析以及乔瑜等[14]、侯金丽等[12]对CH4氧化的相关研究,绘制关键自由基OH·,H·作用于甲烷氧化过程示意图,如图3所示。
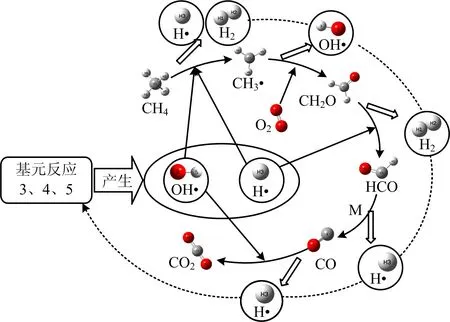
图3 H·与OH·自由基对甲烷氧化主要路径的作用
由图3可知,当甲烷与氧气充足时,甲烷的主要氧化路径为CH4→CH3·→CH2O→HCO→CO→CO2。OH·参与CH4→CH3·和CO→CO22个阶段;H·则作用于CH4→CH3·与CH2O→HCO;O·没有直接参与甲烷的氧化,而是作用于基元反应3,4,不断为甲烷氧化提供H·,OH·;此外,甲烷在氧化过程中生成的H2,OH·,H·又不断促进基元反应3,4,5将H2与O2转化为H·,OH·,形成1个正反馈。
2.2 甲烷爆炸微观反应热力学及动力学分析
运用DFT理论B3LYP-D3(BJ)/6-31+G*水平对构型优化得到的稳定结构进行频率计算,并在M06-2X/def2-TZVPP水平下计算电子能量E,计算条件为常温常压,通过式(1)~(3)计算得到CH4爆炸反应机理中各反应的焓变ΔHΘ、吉布斯自由能变ΔGΘ、正逆反应自由能垒ΔG≠,如表2所示。
分析表2可知,反应1 CH3·+H·→CH4的ΔHΘ=-433.7 kJ·mol-1<0,放热量大,ΔGΘ<0,常温常压下反应可自发进行,CH3·与H·结合生成CH4,一定程度上延缓CH4的氧化进程。反应2 CH4+H·→CH3·+H2,ΔHΘ=1.6 kJ·mol-1>0,吸热,ΔGΘ<0,高温条件下反应自发进行,从动力学的角度上分析ΔGa≠(67.7 kJ·mol-1)<ΔGb≠(70.9 kJ·mol-1),即反应更利正向进行,消耗H·将CH4氧化生成CH3·和H2。
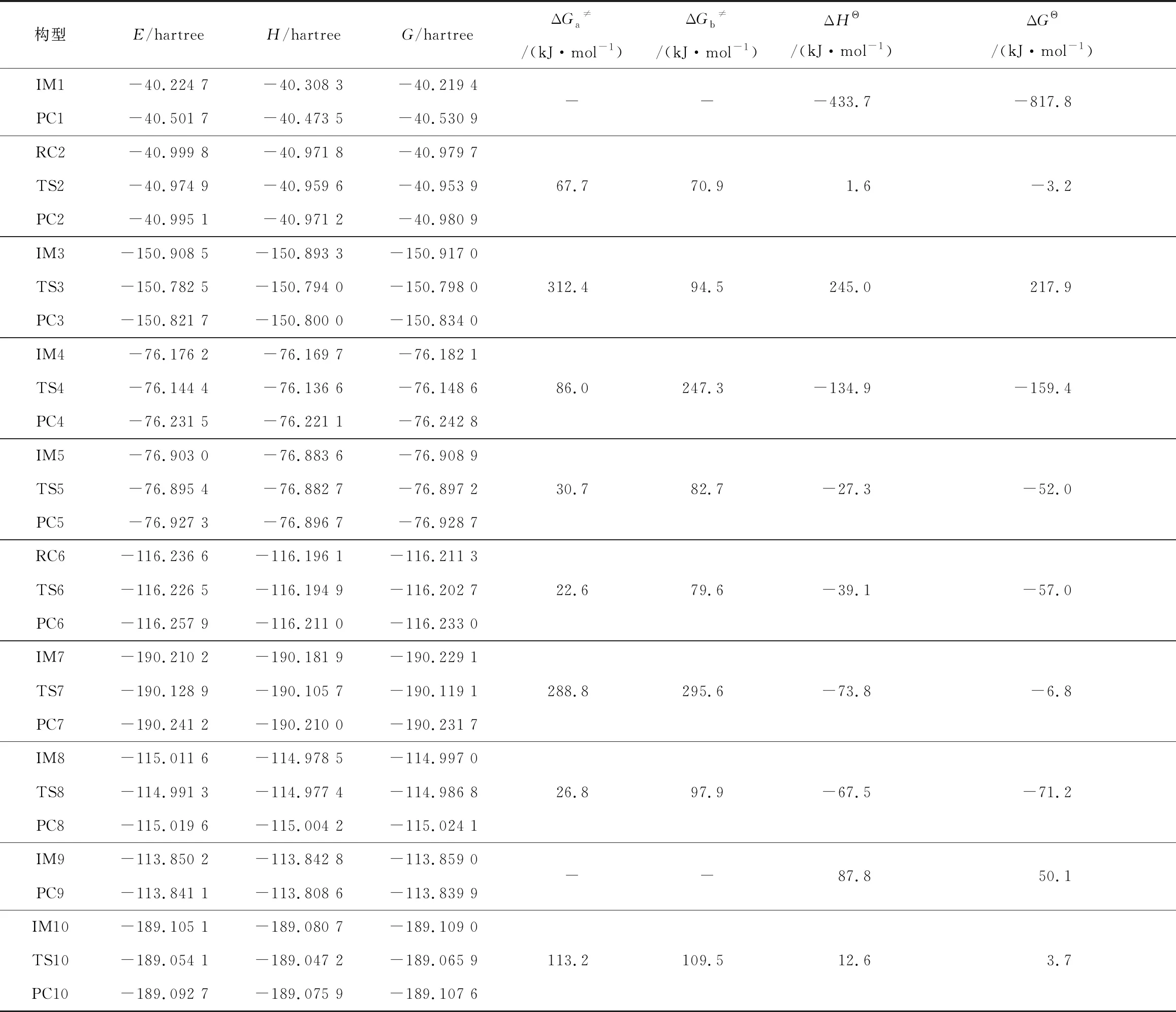
表2 CH4爆炸微观反应机理各基元反应热力学数据及自由能垒
众多研究表明OH·和H·是甲烷爆炸反应机理中起重要作用的关键基团。·OH是羟基(OH-)失去了1个电子的中性形式,具有极强的氧化性。罗振敏等[17]研究发现NH3有与H·结合的趋势,最终通过消耗OH·来实现惰化甲烷的目的。贾海林等[26]研究中发现氢氧化钠通过捕获甲烷爆炸过程中的OH·和H·来实现抑爆。以下对OH·的生成展开详细分析:反应3 O2+H·→OH·+O·反应吸热,ΔHΘ=245.0 kJ·mol-1>0,ΔGΘ>0,常温常压条件下反应非自发进行,从动力学的角度分析ΔGa≠(312.4 kJ·mol-1)>ΔGb≠(94.5 kJ·mol-1),更利于反应逆向进行,消耗OH·和O·生成O2和H·。反应4 O·+H2→OH·+H·的ΔHΘ=-134.9 kJ·mol-1<0,反应放热较大,ΔGΘ<0,常温常压条件下可自发反应,从动力学的角度分析ΔGa≠(86.0 kJ·mol-1)<ΔGb≠(247.3 kJ·mol-1),反应更利于正向进行,消耗O·将H2转化为OH·和H·。对比反应3,4的热力学及动力学数据发现,在反应初期氧气充足,会阻碍反应3的逆反应自发进行,其次,反应4的ΔGa≠(86.0 kJ·mol-1)<反应3的ΔGb≠(94.5 kJ·mol-1),即同样条件下反应4正反应先发生,不断生成OH·和H·并放热。H·的不断生成增加了反应3的中H·浓度,从更而有利于反应3正向反应生成OH·和O·,进一步为甲烷氧化提供OH·,同时反应3生成的O·提高了反应4中O·的浓度,再次促进反应4正向反应。这与侯金丽等[12]基于敏感性分析方法得出反应3和4一起将O2和H2转化为2个OH·的结果一致。
反应5 OH·+H2→H2O+H·的ΔGΘ<0、ΔHΘ=-27.3 J/mol<0,得出:反应放热,正反应在常温常压条件下自发,从动力学角度分析ΔGa≠(30.7 kJ·mol-1)< ΔGb≠(82.7 kJ·mol-1),反应更利于正向进行。反应6 CH4+OH·→CH3·+H2O的ΔGΘ<0,ΔHΘ=-39.1 kJ·mol-1<0,得出:反应6放热,在常温常压条件下可自发进行,ΔGa≠(22.6 kJ·mol-1)<ΔGb≠(79.6 kJ·mol-1),从动力学角度分析反应6更利于正向进行,消耗OH·将CH4氧化为CH3·和H2O,与姜海洋等[24]在M062X/6-31+G(2df,p)水平下计算得到的趋势相同。反应7 CH3·+O2→CH2O+OH·的ΔHΘ=-73.8 kJ/mol<0,放热,ΔGΘ<0,常温常压条件下反应可自发进行,ΔGa≠(288.8 kJ·mol-1)< ΔGb≠(295.6 kJ·mol-1),更利于反应正向进行,氧化CH3·生成CH2O和OH·,同时增加OH·浓度,加速反应进程,姜海洋等[24]在M062X/6-31+G(2df,p)水平下分析反应7所得的热力学及动力学趋势一致。反应8 CH2O+H·→HCO·+H2的ΔHΘ=-67.5 J/mol<0,放热,ΔGΘ<0,常温常压条件下反应可自发进行,ΔGa≠(26.8 kJ·mol-1)< ΔGb≠(97.9 kJ·mol-1),从动力学角度分析反应8更利于反应正向进行,消耗H·将CH2O进一步氧化脱氢,生成HCO·和H2,加速CH4氧化进程。反应9 HCO·+M→CO+H·+M的ΔHΘ=87.8 J/mol>0,为吸热反应,ΔGΘ>0,在常温常压条件下非自发进行。反应10 CO+OH·→CO2+H·吸热,ΔHΘ=12.6 J/mol>0,ΔGΘ>0,常温常压条件下非自发进行,ΔGa≠(113.2 kJ·mol-1)> ΔGb≠(109.5 kJ·mol-1),更利反应逆向进行,不利于CO氧化为CO2和CH4完全氧化。
分析CH4爆炸反应机理中各基元反应的自由能垒,得出:(R3)312.4 kJ·mol-1>(R7)295.6 kJ·mol-1>(R4)247.3 kJ·mol-1>(R10)113.2 kJ·mol-1>(R8)97.9 kJ·mol-1>(R6)79.6 kJ·mol-1>(R2)70.9kJ·mol-1>(R5)82.7 kJ·mol-1,反应3的正反应自由能垒ΔGa≠最大,为该CH4爆炸微观反应机理的决速步,对应的自由能垒为312.4 kJ·mol-1。CH4爆炸微观反应机理热力学及动力学分析结果,如表3所示。

表3 CH4爆炸微观反应机理热力学及动力学过程
分析表3可知,在常温常压条件下,其中反应1,4,5,6,7,8是放热反应,且反应自发正向进行,共同为反应体系提供热量以保证CH4氧化的不断进行,其中反应1 CH3·+H·→CH4放热最大,ΔHΘ=-433.7 kJ·mol-1;反应3 ΔGa≠最大,是该甲烷爆炸机理的决速步,ΔGa≠=312.4 kJ·mol-1。
3 结论
1)CH4链式爆炸微观反应机理中基元反应1和基元反应9无过渡态,基元反应2,3,4,5,6,7,8,10存在过渡态。
2)基元反应1,4,5,6,7,8的ΔHΘ<0,为放热反应,共同为反应体系供热,保证CH4氧化反应的不断进行,其中基元反应1 CH3·+H·→CH4放热量最大,ΔHΘ=-433.7 kJ·mol-1。
3)从热力学及动力学角度分析得出关键自由基OH·是由基元反应3 O2+H·→OH·+O·与基元反应4 O·+H2→OH·+H·相互协同,互相促进而生成,同时加速了CH4的氧化进程。
4)基元反应3 O2+H·→OH·+O·为该CH4爆炸微观反应机理的决速步,相应的自由能垒为312.4 kJ·mol-1,并且该反应从热力学分析,反应吸热且常温常压条件下逆反应可自发进行,从动力学角度分析,反应利于逆向进行。
猜你喜欢
四川大学学报(自然科学版)(2022年4期)2022-07-22
北京航空航天大学学报(2022年5期)2022-06-06
兵工学报(2022年2期)2022-05-22
原子与分子物理学报(2022年3期)2022-03-05
兵工学报(2021年4期)2021-06-19
兵工学报(2020年12期)2020-02-06
青岛大学学报(工程技术版)(2019年2期)2019-09-10
科学导报(2018年30期)2018-05-14
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
