
3-苯基-2,3-二氢-1H-苯并[f]吲哚-4,9-二酮的合成研究*
2022-08-02 05:38:04徐小娜
化学工程师 2022年7期
于 雪,徐小娜,柯 苗
(咸阳职业技术学院 医药化工学院,陕西 咸阳 712000)
吲哚是一类非常重要的含氮杂环化合物,吲哚衍生物广泛的存在于天然产物(如马钱子碱、麦角碱、长春碱和长春新碱等)[1-4]及上市药物分子中(如镇定剂利血平、治疗偏头痛的普坦类药物等)[5-7]。因此,研究吲哚衍生物的合成方法具有重要意义。作为吲哚衍生物中具有代表性的一类,苯并吲哚衍生物的合成及应用也得到了广泛的关注[8-11]。但关于苯并[f]吲哚-4,9-二酮衍生物的文献报道较少,其中,2013年,Sun等[12]报道用铜(II)催化萘醌与β-烯胺酮反应,合成了苯并[f]吲哚-4,9-二酮,反应过程中铜(II)盐作为Lewis酸和氧化催化剂。2020年,Christodoulou等[13]报道以含有1,4-萘醌基团的4,4-二取代的4H-异恶唑-5-酮在铑催化剂存在下经过脱羧开环/闭环反应转化成苯并[f]吲哚-4,9-二酮。目前,苯并[f]吲哚-4,9-二酮衍生物的种类还是比较少,因此,继续研发该类化合物的合成方法仍具有重要的研究价值。
为了研发一种3-芳基-2,3-二氢-1H-苯并[f]吲哚-4,9-二酮衍生物的合成方法,进而研究该类化合物的生物活性,本研究报道了3-芳基-2,3-二氢-1H-苯并[f]吲哚-4,9-二酮(1)的合成。以2-羟基萘-1,4-二酮(2)和硝基苯乙烯衍生物(3)为原料,经过Michael加成、硝基还原并关环,两步反应合成目标化合物(1),合成路线见图1。

图1 3-苯基-2,3-二氢-1H-苯并[f]吲哚-4,9-二酮(1a)的合成路线Fig.1 Synthetic route for 3-phenyl-2,3-dihydro-1H-benzo[f]indole-4,9-dione(1a)
1 实验部分
1.1 原料与仪器
2-羟基萘-1,4-二酮(纯度99%)、(E)-硝基苯乙烯(纯度98%)(3a)、1-(3-甲氧苯基)-2-硝基乙烯(纯度98%)(3b)、10%Pd/C(湿基),阿拉丁生物科技有限公司;柱层析硅胶(300~400目,青岛海洋化工厂);其他所有试剂均为市售分析纯。
AV400型核磁共振仪(CDCl3为溶剂,TMS为内标,德国Bruker公司);Ultima Global Spectrometer型质谱仪(ESI源,美国Waters公司);RE-52AA型旋转蒸发仪(上海亚荣生化仪器厂);SHB-Ш型循环水式多用真空泵(郑州长城工贸有限公司)。
1.2 合成方法
1.2.1 中间体(4)的合成 2-羟基萘-1,4-二酮(2)(5mmol)和β-硝基烯3(5.5mmol)溶于二氯甲烷(10mL),搅拌均匀后滴加三乙胺(5mmol),然后室温反应1h,TLC检测反应结束后,浓缩反应液,硅胶柱层析分离(洗脱剂:V石油醚∶V乙酸乙酯=5∶1)。
2-羟基-3-(2-硝基-1-苯基乙基)萘-1,4-二酮(4a):黄色固体,收率98.6%。1H NMR(400MHz,CDCl3)δ:8.11(d,J=7.6Hz,1H),8.06(d,J=7.5Hz,1H),7.76(t,J=7.5Hz,1H),7.68(t,J=7.3Hz,1H),7.47(d,J=7.5Hz,2H),7.32(t,J=7.4Hz,2H),7.28~7.24(m,1H),5.48(dd,J=13.3,9.1Hz,1H),5.34~5.28(m,1H),5.15(dd,J=13.3,6.8Hz,1H)。13C NMR(100MHz,CDCl3)δ:183.73,181.17,153.26,137.55,135.47,133.33,132.64,129.01,128.30,127.87,127.24,126.37,120.83,76.38,39.68。ESI-MS[M+1]+m/z 324.12。
2-羟基-3-[1-(3-甲氧基苯基)-2-硝基乙基]萘-1,4-二酮(4b):黄色固体,收率92.1%。1H NMR(400MHz,CDCl3)δ:8.11(d,J=7.6Hz,1H),8.06(d,J=7.6Hz,1H),7.79~7.74(m,1H),7.68(t,J=7.1Hz,1H),7.23(t,J=8.0Hz,1H),7.04(dd,J=11.7,4.9Hz,2H),6.79(dd,J=8.2,2.1Hz,1H),5.47(dd,J=13.4,9.0Hz,1H),5.32~5.25(m,1H),5.14(dd,J=13.4,6.8Hz,1H),3.78(s,3H)。13C NMR(100MHz,CDCl3)δ:183.69,181.16,159.91,153.29,138.99,135.45,133.32,132.64,129.99,129.02,127.25,126.36,120.71,120.54,114.38,112.94,76.32,55.26,39.63。ESI-MS[M+1]+m/z 354.13。
1.2.2 目标化合物(1)的合成 N2保护下,将中间体4(0.5mmol)溶于甲醇(8mL),搅拌均匀后加入10%的Pd/C(20mg),然后用H2置换体系中的N23次,随后在H2氛围下反应8h,TLC检测反应结束后,减压抽滤,滤液减压浓缩,粗品经硅胶柱层析分离纯化(洗脱剂:V石油醚∶V乙酸乙酯=5∶1)。
3-苯基-2,3-二氢-1H-苯并[f]吲哚-4,9-二酮(1a):紫色固体,收率43.3%。1H NMR(400MHz,CDCl3)δ:8.00(d,J=1.1Hz,1H),7.98(d,J=1.1Hz,1H),7.65(td,J=7.6,1.1Hz,1H),7.56(td,J=7.5,1.1Hz,1H),7.37~7.27(m,4H),7.25~7.18(m,1H),5.24(brs,1H),4.69(dd,J=11.8,5.9Hz,1H),4.24~4.15(m,1H),3.80~3.72(m,1H)。13C NMR(100MHz,CDCl3)δ:180.33,179.59,153.20,142.97,134.63,134.53,131.65,131.58,128.81,127.25,127.08,126.02,125.62,121.22,55.35,46.31。ESI-MS[M+1]+m/z 276.13。
3-(3-甲氧基苯基)-2,3-二氢-1H-苯并[f]吲哚-4,9-二酮(1b):淡红色固体,收率为47.1%。1H NMR(400MHz,CDCl3)δ:8.01(d,J=1.1Hz,1H),7.98(d,J=1.1Hz,1H),7.66(td,J=7.6,1.1Hz,1H),7.56(td,J=7.5,1.1Hz,1H),7.33~7.27(m,1H),7.24~7.16(m,3H),7.11~7.05(m,1H),5.21(brs,1H),4.67~4.61(m,1H),4.27~4.19(m,1H),3.81(s,3H),3.84~3.75(m,1H)。13C NMR(100MHz,CDCl3)δ:181.72,178.44,156.29,147.54,138.65,134.76,131.43,131.02,128.85,127.56,127.11,126.32,123.65,119.42,77.19,56.89,47.50。ESI-MS[M+1]+m/z 306.16。
2 结果与讨论
2.1 中间体(4a)的合成
在三乙胺碱性条件下,2-羟基萘-1,4-二酮(2)和(E)-硝基苯乙烯(3a)发生Michael加成反应制备中间体2-羟基-3-(2-硝基-1-苯基乙基)萘-1,4-二酮(4a),反应中溶剂、物料比、反应时间对产物收率具有重要影响,考察结果见表1。

表1 反应条件对中间体4a收率的影响Tab.1 Effect of reaction conditions on the yield of compound 4a
首先,固定物料比n3a∶n2=1.0∶1、反应时间2h,在该条件下考察反应溶剂对中间体4a收率的影响。
2.1.1 溶剂的选择 由表1(Entry 1,2,3,4)发现,反应在4种溶剂中均可以进行,而在二氯甲烷中反应,中间体4a收率最高,达到92.5%(Entry 4),因此,确定适宜的反应溶剂为二氯甲烷。
2.1.2 物料比的确定 在二氯甲烷溶剂中,固定反应时间为2h,考察物料比的影响。由表1(Entry 4,5,6)发现,考虑到后处理难易程度,选择化合物3a过量(硝基烯3a与产物极性差异大,化合物2与产物极性接近,反应结束后柱层析纯化,过量的3a容易除去),发现增加3a的用量,当物料比n3a∶n2=1.1∶1时,产物收率明显增加到98.6%(Entry 5),而继续增加3a用量为n3a∶n2=1.2∶1时,产物收率没有明显变化,因此,选择适宜的物料比为n3a∶n2=1.1∶1。
2.1.3 反应时间的确定 在确定好溶剂和物料比后,考察反应时间(Entry 6,7,8,9),发现缩短反应时间为1h,中间体4a收率明显降低为83.9%,而增加反应时间为3和4h,中间体4a收率没有明显增加,因此,确定适宜的反应时间为2h。
最终确定该Michael加成反应的最佳反应条件为:反应溶剂为二氯甲烷;物料比为n3a∶n2=1.1∶1;反应时间为2h。
2.2 产物(1a)的合成
中间体2-羟基-3-(2-硝基-1-苯基乙基)萘-1,4-二酮(4a)在Pd/C加氢条件下经历两步反应过程,首先是硝基被还原为伯胺,由于萘醌结构上的2-位羟基可以发生酮与烯醇的互变,然后伯胺作为亲核试剂进攻羰基碳,随后经历脱水过程,得到目标产物3-苯基-2,3-二氢-1H-苯并[f]吲哚-4,9-二酮(1a)。该还原反应过程中使用Pd/C,具有一定的危险性,所以首先需要注意加料顺序,加入反应物和溶剂后再加入Pd/C,此过程需要在N2保护下进行。加完Pd/C后,撤去N2,然后用H2置换3次,目的是除去体系中的N2和溶剂中溶解的少量空气,从而增加还原反应的效率。
在该还原反应过程中,反应时间是影响产物(1a)收率的主要因素,对反应时间的考察结果见表2。

表2 反应时间对产物(1a)收率的影响Tab.2 Effect of reaction time on the yield of product(1a)
由表2可知,当反应时间为5h,产物(1a)收率只有21.6%(Entry 1),增加反应时间,产物收率明显提高,当反应时间为8h时,收率提高至43.3%(Entry 3),继续延长反应时间,收率没有明显变化(Entry 4,5)。因此,确定最佳反应时间为8h。
2.3 中间体(4a)及产物(1a)结构表征分析
对中间体(4a)的1H NMR进行分析(图2):δ 8.11处双重峰、δ8.06处双重峰、δ7.76处三重峰、δ 7.68处三重峰、δ7.47处双重峰、δ7.32处三重峰,δ 7.28~7.24处三重峰,峰面积积分合计为9H,归属为芳香氢;δ5.48处,dd峰,偶合常数分别为J=13.3Hz、9.1Hz,积分为1H,归属为硝基相连接的亚甲基氢;δ 5.34~5.28,多重峰,积分为1H,归属为苯环连接的次甲基氢;δ5.15处,dd峰,偶合常数分别为J=13.3Hz、6.8Hz积分为1H,归属为硝基相连接的亚甲基氢。对中间体(4a)的13C NMR进行分析(图3):δ183.73和181.17为萘醌结构上的两个羰基碳信号;δ153.26,137.55,135.47,133.33,132.64,129.01,128.30,127.87,127.24,126.37,120.83,均为萘醌和苯环碳信号;δ 76.38为与硝基相连接的亚甲基碳信号;δ39.68为与苯环相连接的次甲基碳信号。再结合质谱数据进一步证明结构的正确性。

图2 中间体(4a)的核磁共振氢谱Fig.2 1H NMR of compound(4a)

图3 中间体(4a)的核磁共振碳谱Fig.3 13C NMR of compound(4a)
对产物(1a)的1H NMR进行分析(图4):δ8.00处d峰、δ7.98处d峰、δ7.65处td峰、δ7.56处td峰、δ7.37~7.27处多重峰、δ7.25~7.18处多重峰,峰面积积分合计为9H,归属为芳香氢;δ5.24处宽峰,积分1H,归属为吡咯NH;δ4.69处的dd峰,偶合常数分别为J=11.8,5.9Hz,积分为1H,归属为吡咯亚甲基氢;δ4.24~4.15处多重峰,积分为1H,归属为与苯环相连接的次甲基氢;δ3.80~3.72处多重峰,积分为1H,归属为吡咯亚甲基氢。对产物(1a)的13C NMR进行分析(图5):δ180.33和179.59为萘醌结构上的两个羰基碳信号;δ153.20,142.97,134.63,134.53,131.65,131.58,128.81,127.25,127.08,126.02,125.62,121.22,均为萘醌和苯环碳信号;δ55.35为吡咯亚甲基碳信号;δ46.31为与苯环相连接的次甲基碳信号。

图4 产物(1a)的核磁共振氢谱Fig.4 1H NMR of target compound(1a)
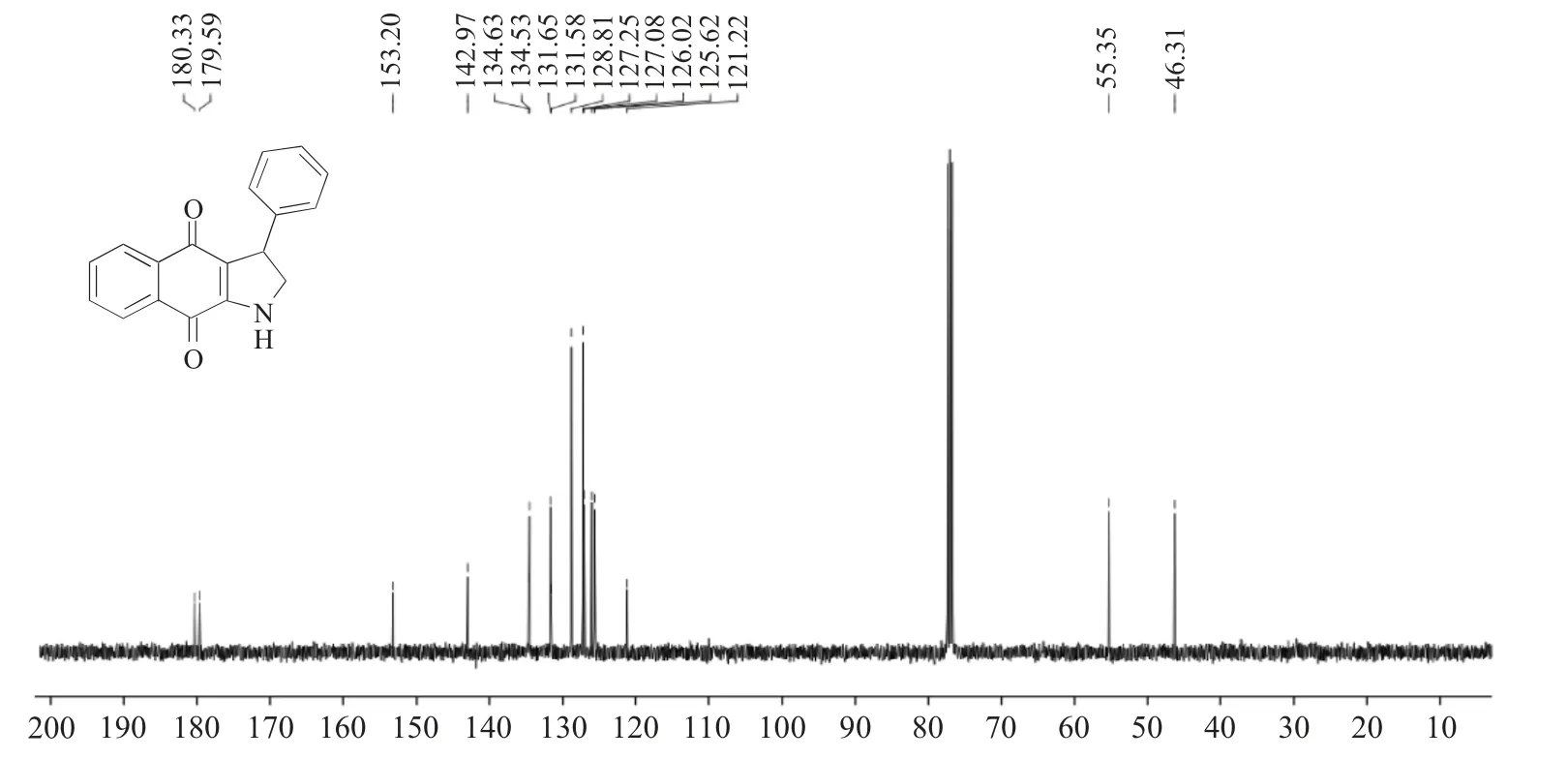
图5 产物(1a)的核磁共振碳谱Fig.5 13C NMR of target compound(1a)
2.4 反应底物拓展
对于反应底物拓展研究发现,在最佳反应条件下,2-羟基萘-1,4-二酮(2)和1-(3-甲氧苯基)-2-硝基乙烯(3b)发生Michael加成反应得到2-羟基-3-[1-(3-甲氧基苯基)-2-硝基乙基]萘-1,4-二酮(4b),收率为92.1%。中间体(4b)再经过Pd/C加氢反应,得到3-(3-甲氧基苯基)-2,3-二氢-1H-苯并[f]吲哚-4,9-二酮(1b),收率为47.1%。
3 结论
在三乙胺碱性条件下,2-羟基萘-1,4-二酮(2)和(E)-硝基苯乙烯(3a)发生Michael加成反应制备中间体2-羟基-3-(2-硝基-1-苯基乙基)萘-1,4-二酮(4a),再经过Pd/C加氢反应,得到目标化合物3-苯基-2,3-二氢-1H-苯并[f]吲哚-4,9-二酮(1a),并分别对两步反应条件进行了考察,确定该Michael加成反应的适宜条件为:溶剂为二氯甲烷,物料比为n3a∶n2=1.1∶1,反应时间为2h,还原反应时间为8h。对该反应工艺的底物拓展发现,1-(3-甲氧苯基)-2-硝基乙烯(3b)同样适用于该反应条件,可以得到较高收率的产物。该合成方法为3-芳基-2,3-二氢-1H-苯并[f]吲哚-4,9-二酮的合成提供了可行的方法,并为苯并[f]吲哚母核活性化合物的开发提供大量结构新颖的化合物分子,具有较高的应用价值。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13 06:40:42
昆明医科大学学报(2020年12期)2021-01-26 00:44:12
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
山东化工(2019年11期)2019-06-26 03:26:44
国际呼吸杂志(2019年1期)2019-03-08 03:07:02
杭州师范大学学报(自然科学版)(2016年2期)2016-05-05 03:22:14
绥化学院学报(2016年3期)2016-03-23 09:00:14
西北师范大学学报(自然科学版)(2015年6期)2016-01-19 01:55:14
无机化学学报(2014年12期)2014-02-28 17:34:01
