
育龄期21-羟化酶缺乏症误诊为多囊卵巢综合征二例并文献复习
2022-08-02 13:27迟学秀郭宝强于姗何东华
国际生殖健康/计划生育杂志 2022年4期
迟学秀,郭宝强,于姗,何东华
先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH)是一组由于肾上腺皮质类固醇激素合成过程中酶的缺陷所引起的常染色体隐性遗传病,其中21-羟化酶缺乏症(21-hydroxylase deficiency,21-OHD)最常见,主要由CYP21A2基因缺陷引起,约占CAH的90%~95%,文献报道21-OHD发病率约1/20 000~1/10 000,根据基因型与临床表型的关系,21-OHD分为经典型和非经典型,前者又分为失盐(salt-wasting)型和单纯男性化(simple virilizing)型[1-2]。经典型21-OHD患者临床症状典型、诊断容易且不易误诊,非经典型21-OHD患者临床症状较轻且不典型,与多囊卵巢综合征(polycystic ovarian syndrome,PCOS)在临床表现、实验室检查、影像学检查等多方面均有重叠,且由于PCOS发病率高及缺乏特异性的诊断标准,非经典型21-OHD临床中易误诊为PCOS,这已成为临床比较突出的一个问题,治疗延迟会严重影响患者的生长发育及生育功能。据报道,非经典型21-OHD国外发病率可达1/1 000~1/200[3],且与种族及地域有关。现分析2例曾误诊为PCOS的育龄期21-OHD患者的临床资料及基因检测结果,总结其诊疗思路并复习相关文献,为临床工作者对21-OHD育龄期女性的诊疗提供参考,减少漏诊及误诊。
1 病例报告
1.1 病例1患者女,29岁,因月经紊乱十余年,婚后未避孕未孕5年,于2020年6月15日就诊于聊城市第二人民医院(我院)内分泌科。个人史:足月产,出生时无外生殖器异常,智力正常,父母非近亲结婚。月经史:12岁初潮,月经不规律,经期5~7 d,月经周期20~60 d。否认遗传疾病家族史。患者反复睾酮升高,介于3.07~3.83 nmol/L(正常值0.29~1.67 nmol/L)十余年,未予重视。婚后4年未避孕一直未孕,2019年10月患者因不孕到外院妇科门诊就诊,妇科超声提示多囊样卵巢,诊断为PCOS,行促排卵治疗,效果不佳,后因睾酮一直高于正常且未孕就诊于我院内分泌科。入院查体:身高153 cm,体质量53 kg,血压116/76 mmHg(1 mmHg=0.133 kPa),面部皮肤油腻,可见痤疮,上唇小胡须,甲状腺无肿大,腹部可见散在毛发,背部可见少许毛发,心肺查体未见异常,改良Ferriman-Gallwey评分9分(评分≥5分诊断为多毛症),双乳房TannerⅤ期,女性外生殖器无畸形,阴毛呈菱形分布,TannerⅤ期,四肢肌肉健壮。染色体核型分析正常(46,XX),2020年6月16日卵泡期内分泌相关指标见表1。黄体生成素(luteinizing hormone,LH)/卵泡刺激素(follicle-stimulating hormone,FSH)比值2.52,血清皮质醇、促肾上腺皮质激素、生长激素、促甲状腺激素、血糖、胰岛素、肾素和醛固酮水平均正常。肾上腺增强CT提示左侧肾上腺内侧支略增粗,垂体增强磁共振成像(magnetic resonance imaging,MRI)未见异常。基因测序发现患者存在2个位点杂合突变,第125位氨基酸由精氨酸变为组氨酸(p.Arg125His,c.374G>A,Het,见图1A)和第173位氨基酸由异亮氨基酸变为天冬酰胺(p.Ile173Asn,c.518T>A,Het,见图1B);患者之母存在c.518T>A(p.Ile173Asn)杂合错义突变(见图1C);患者之父存在c.374G>A(p.Arg125His)杂合错义突变(见图1D)。诊断为CAH(非经典型21-OHD)。于2020年6月21日给予患者每晚睡前0.375 mg地塞米松,密切随访患者内分泌相关指标(见表1)。1个月后患者痤疮改善,面部皮肤变细腻,月经规律来潮,睾酮水平降至正常。2个月后患者自然妊娠,将地塞米松改为早晚各口服10 mg氢化可的松,患者于2021年5月25日顺产一男婴。
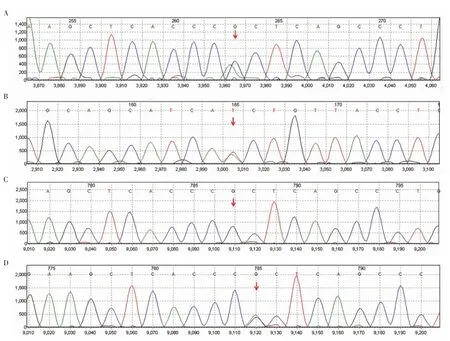
图1 病例1及其亲属CYP21A2基因测序结果

表1 病例1不同时间各项激素水平变化情况
1.2 病例2患者女,27岁,因月经紊乱9年,婚后未避孕未孕3年,于2020年8月16日就诊于我院内分泌科。个人史:足月产,出生时无外生殖器异常,智力正常,父母非近亲结婚。月经史:18岁初潮,月经不规律,经期5~8 d,月经周期20~37 d。否认遗传疾病家族史。9年前患者月经初潮后因周期不规律,曾行中药治疗,效果欠佳。婚后3年自然妊娠2次,均于妊娠2个月时无明显诱因流产,2019年8月因不孕到外院妇科门诊就诊,查睾酮70.89 nmol/L,妇科超声提示多囊样卵巢,肾上腺CT示双侧肾上腺增生,染色体核型分析正常(46,XX)。诊断为PCOS,给予促排卵药物治疗,效果不佳,为进一步治疗就诊于我院内分泌科。入院查体:身高156 cm,体质量57.5 kg,体质量指数23.63 kg/m2,血压126/78 mmHg,神志清,女性男性化,上唇小胡须,甲状腺不大,心肺查体未见异常,胸腹部可见散在毛发,改良Ferriman-Gallwey评分9分,双乳房TannerⅢ期,阴蒂肥大,阴毛呈倒三角形分布,TannerⅤ期。2020年8月17日卵泡期内分泌相关指标见表2。LH/FSH比值0.17,血清皮质醇、促肾上腺皮质激素、雌二醇、胰岛素、血糖、肾素和醛固酮水平均正常。基因测序发现患者存在1个位点纯合突变,第173位氨基酸由异亮氨基酸变为天冬酰胺(p.Ile173Asn,c.518T>A,Hom,见图2),未能采集到患者双亲的血样进行检测。诊断为CAH(单纯男性化型21-OHD)。于2020年8月21日每晚睡前给予0.375 mg地塞米松,规律随访患者内分泌相关指标(见表2),治疗1个月后患者体毛改善,月经规律来潮,目前随访至2021年1月患者未避孕未孕。
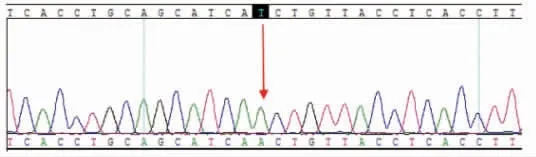
图2 病例2 CYP21A2基因测序结果
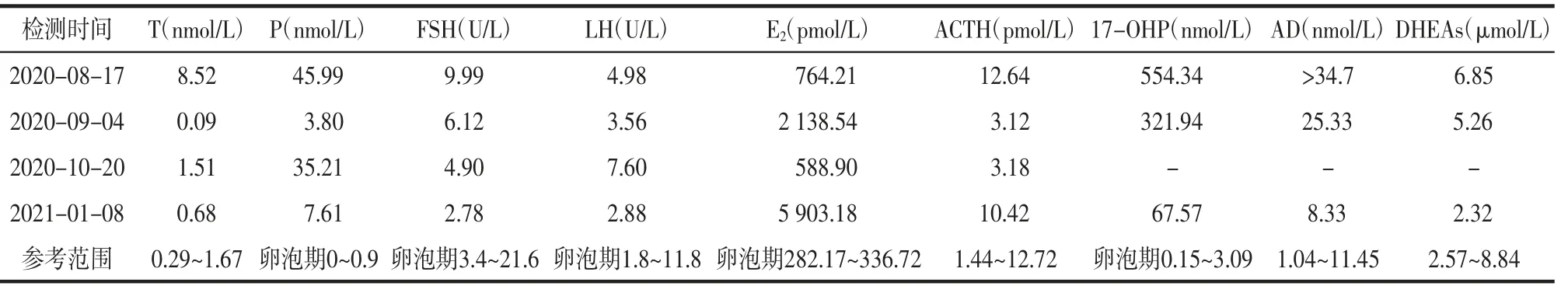
表2 病例2不同时间段各项激素水平的变化
2 讨论
2.1 21-OHD的发病机制21-OHD是由CYP21A2基因缺陷导致21-羟化酶(P450c21)功能不足所致的隐性遗传疾病。该基因位于6p21.3,与无活性的假基因CYP21AIP相邻,具有98%相同的外显子序列[4],两个基因高度同源且排列位置相近,该区基因极易发生基因重组或转换,导致CYP21A2基因缺陷。目前已发现200余种CYP21A2基因突变类型,大约70%的CYP21A2基因突变由于假基因的微转换,25%~30%由于大片段的缺失和基因嵌合,只有1%~2%的突变来自CYP21A2基因的新突变[5]。CYP21A2基因突变和临床表型关系密切,酶活性缺失与临床表型有较高的一致性,其中失盐型21-OHD一致性达100%,单纯男性化型21-OHD一致性达95%,非经典型21-OHD一致性达70%[6]。研究显示国内失盐型、单纯男性化型和非经典型21-OHD中最常见的突变分别是基因片段缺失、I173N和P31L点突变[7-8]。不同地区、种族的CYP21A2基因突变存在差异,国内一项研究发现徐州地区21-OHD患儿常见基因变异位点为c.293-13C>G、c.518T>A和Exonl-7del[9],天津及周边地区21-OHD患儿常见基因变异分别为c.293-13C/A>G(12G)、大片段基因缺失或转换和I173N点突
变[10]。基因检测是CAH诊断的金标准,临床疑似或生化诊断困难者,建议常规行基因突变筛查[11]。本研究2例患者均通过基因检测最终分别明确诊断为非经典型21-OHD和单纯男性化型21-OHD。其中p.Ile173Asn和p.Arg125His两个位点为既往报道过的有意义的突变[11-13],可导致21-羟化酶活性下降。
2.2 21-OHD的诊断及与PCOS的鉴别诊断21-OHD诊断主要依据临床表现、生化检查和激素检测、家族史及基因检测。因为21-OHD的酶缺陷程度不同,不同年龄女性的临床表现存在差异。临床中经典型21-OHD诊断相对容易,非经典型21-OHD女性患者主要以月经紊乱、不孕及高雄激素血症为特征,其临床表现与PCOS相似,将非经典型21-OHD误诊为PCOS已成为一个较为突出的临床问题。由于非经典型21-OHD患者21-羟化酶活性保留20%~50%,皮质醇及醛固酮分泌大多正常,临床主要为不同程度的高雄激素血症表现[13]。在高水平雄激素的长期作用下,促性腺激素的释放受到干扰,LH产生增多,可导致多囊样卵巢,非经典型21-OHD患者约40%伴有多囊样卵巢[14]。而两种疾病治疗方案截然不同,故鉴别诊断十分重要。二者鉴别诊断主要依靠检测17-羟孕酮水平、促肾上腺皮质激素兴奋试验及基因检测。近年国内有报道显示21-OHD和PCOS的最佳鉴别指标为17-羟孕酮和孕酮,最佳截断值分别为10.02 nmol/L和1.99 nmol/L[15]。并且提出17-羟孕酮可作为21-OHD一级筛查指标,但其可能受应激、胎龄、疾病状态等影响,因此应注意筛查结果会呈现假阳性[16]。基础17-羟孕酮>30 nmol/L(1 000 ng/dL)考虑为经典型21-OHD,基础17-羟孕酮<6 nmol/L(200 ng/dL)考虑为非经典型21-OHD,基础17-羟孕酮为6~30 nmol/L(200~1 000 ng/dL)需要进行促肾上腺皮质激素兴奋试验,再次检测17-羟孕酮>30 nmol/L则考虑为经典型21-OHD,若17-羟孕酮<30 nmol/L则考虑可能存在非经典型21-OHD[16]。国外既往报道PCOS患者肾上腺呈高反应状态,促肾上腺皮质激素刺激后也会出现17-羟孕酮和皮质醇升高,但17-羟孕酮一般低于16 noml/L,而非经典型21-OHD患者17-羟孕酮可高于45 noml/L[17]。然而国内部分医院未开展17-羟孕酮检测,或由于各种原因不能获得促肾上腺皮质激素药物,导致未开展促肾上腺皮质激素兴奋试验。而大多数医院可常规检测孕酮,通过皮质醇产生途径可知21-羟化酶缺乏时17-羟孕酮和孕酮水平都会升高。既往研究发现卵泡期显著增高的孕酮水平对非经典型21-OHD和PCOS初步鉴别诊断有一定提示意义,非经典型21-OHD患者的孕酮水平多超过排卵后孕酮水平即9.54 nmol/L;而PCOS不排卵患者的孕酮水平多处于排卵期前低值[14]。中剂量地塞米松抑制试验也可用于二者的鉴别诊断,非经典型21-OHD组患者17-羟孕酮、孕酮、睾酮和雄烯二酮抑制率显著高于PCOS组,17-羟孕酮、孕酮、睾酮和雄烯二酮抑制率的最佳裁断值分别为73.5%、55.5%、68.3%和61.4%[15]。本研究2例患者以睾酮、孕酮升高作为线索,最终通过检测17-羟孕酮水平及基因检测而确诊。
2.3 21-OHD的治疗21-OHD的治疗主要包括药物和手术治疗。近年研究显示糖皮质激素是21-OHD的一线治疗方法,糖皮质激素主要通过抑制高促肾上腺皮质激素介导的高雄激素血症替代类固醇激素缺失。临床上根据患者类型,可加用盐皮质激素或补充氯化钠来治疗。传统激素治疗在实际临床管理中仍有很多不足,如传统激素难以模拟皮质醇的生理性昼夜节律,使临床管理在达到治疗目的与防止激素过量之间难以权衡。近年来,新的给药模式及补充治疗在21-OHD治疗研究中取得了进展,目的是接近生理性糖皮质激素替代治疗,如氢化可的松口服缓释制剂、氢化可的松泵、儿童剂量的氢化可的松制剂的应用,这可能为21-OHD患者提供更安全有效的治疗[18]。本研究2例患者给予地塞米松抑制过多的促肾上腺皮质激素释放,从而减轻雄激素的过度产生,治疗后2例患者雄激素增多症状明显改善、恢复规律月经,其中病例1成功自然妊娠并顺产一健康男婴。
综上所述,21-OHD临床中并不少见,非经典型21-OHD患者缺乏典型临床症状,临床中易误诊为PCOS从而延误治疗,其明确诊断依赖于17-羟孕酮测定、促肾上腺皮质激素兴奋试验及中剂量地塞米松抑制试验,CYP21A2基因突变检测在21-OHD诊断及遗传咨询中亦有重要作用,临床工作者应提高对21-OHD的认识,避免延误诊治。
猜你喜欢
现代泌尿生殖肿瘤杂志(2022年1期)2022-11-21
江苏卫生保健(2022年5期)2022-01-01
人人健康(2021年13期)2021-11-30
云南医药(2021年3期)2021-07-21
青年时代(2018年24期)2018-10-13
课程教育研究·学法教法研究(2018年22期)2018-08-11
中国体育教练员(2017年3期)2018-01-19
中国体育教练员(2017年2期)2017-07-31
教育(2017年12期)2017-05-04
大众健康(2016年3期)2016-05-31
