
铜死亡与铜代谢相关疾病研究进展
2022-08-01 02:25刘骏达钟薇薇鲁显福李元海
江苏大学学报(医学版) 2022年4期
刘骏达,钟薇薇,鲁显福,李元海
(1. 安徽医科大学第一附属医院高新院区麻醉科,安徽 合肥 230088;2. 安徽医科大学第一附属医院麻醉科,安徽 合肥 230022)
铜是一种常见的金属元素,也是一种过渡元素,具有氧化还原活性。在常规的化学反应和生理条件下,还原型Cu+可转变为氧化型Cu2+[1]。铜离子通过提供或接受电子参与多种生化反应[2]。铜离子可与多种蛋白质或酶结合,作为辅因子或结构组成部分,参与调控能量代谢、线粒体呼吸和抗氧化等多个生理过程。铜离子的含量维持动态平衡,失衡可导致氧化应激[3]和细胞自噬异常[4]等,从而诱发多种铜或铜离子相关性疾病的发生。
2022年3月17日,Tsvetkov等[5]首次提出一种具有铜依赖性的、全新的细胞死亡方式——“铜死亡(cuprotosis)”,其不同于其他已知的细胞死亡方式,如细胞凋亡、焦亡(pyroptosis)和坏死性凋亡(necroptosis),而类似于锌死亡和铁死亡,是一种金属离子诱导的调节性细胞死亡。类似铁死亡[6],Cu2+通过直接与三羧酸循环中脂酰化部分相结合,诱导脂酰化蛋白的聚集和铁-硫簇蛋白的不稳定,导致蛋白毒性应激,从而诱发不依赖于细胞凋亡途径的细胞死亡。本文针对铜死亡的发现及铜相关疾病作一综述。
1 铜代谢的基本过程
正常人体内含铜100~200 mg,50%~70%存在于肌肉及骨骼,20%存在于肝脏,5%~10%分布于血液[7]。人体所需铜主要通过饮食获取,如红肉、牛奶与坚果等。膳食中的铜主要以Cu2+形式存在,但无法被细胞直接利用。经典理论认为,消化道上皮细胞表面存在多种还原酶,可将Cu2+还原为Cu+,然后与铜转运蛋白1(copper transporter 1,CTR1)结合进入肠上皮细胞[7]。Cu+经小肠上皮细胞吸收后由铜转运ATP 酶α(copper-transporting ATPase alpha,ATP7A)释放至门静脉循环中。大部分新吸收的Cu+再经CTR1介导进入肝细胞,其中绝大部分的Cu+通过铜伴侣蛋白抗氧化物-1(antioxidant 1,Atox1)-铜转运ATP 酶β(copper-transporting ATPase beta,ATP7B)-高尔基复合体途径与α2-球蛋白(即前铜蓝蛋白)结合形成铜蓝蛋白,转运至全身各系统。肝细胞内过量的铜经ATP7B以囊泡形式分泌至胆汁中,绝大部分以粪便的形式排出体外,小部分经过消化道再次重吸收[8]。当外周铜浓度下降,ATP7A可以将铜从肝脏储存中调动至血液中,维持外周循环有效的铜浓度。
铜离子的吸收、转运、储存和排泄过程共同决定了铜代谢稳态的调节过程,铜离子含量的过量和缺乏均可导致各种疾病的发生。
2 铜死亡的发现及机制
尽管“铜死亡”的命名于2022年首次提出,但铜死亡的相关研究最初始于2019年。来自MIT和Harvard大学Broad研究所的Todd Golub团队发现了两种可以携带铜离子穿过细胞膜的小分子:双硫仑(disulfiram)和伊利司莫(elesclomol),经证实这两种Cu2+载体可以杀死特定的耐药癌细胞[9]。
双硫仑又名戒酒硫,是一种酒精戒断药物,已经在临床中用于治疗酒精成瘾长达60年。双硫仑的抗肿瘤作用需要Cu2+参与,其在体内的代谢产物二硫代氨基甲酸二乙酯(diethyldithiocarbamate, DTC)与Cu2+形成螯合物——二乙基二硫代氨基甲酸铜[copper(Ⅱ) diethyldithiocarbamate, CuET]。CuET促进核蛋白定位蛋白4(nuclear protein localization protein 4, NPL4)凝结并与p97蛋白紧密结合,影响肿瘤细胞内p97蛋白发挥降解蛋白质的功能,致使大量废弃蛋白在细胞内过度积累,最终导致肿瘤细胞死亡[10]。
伊利司莫由Synta Pharmaceuticals Corp公司研发,既往称作STA-4783,最早于2003年由Berkenblit等在欧洲癌症研究与治疗组织(EORTC)、美国国家癌症研究所(NCI)和美国癌症研究协会(AACR)联合举办的分子靶标与癌症治疗学国际会议上报道[11]。随后多个研究团队开展了一系列针对伊利司莫抗肿瘤的Ⅰ/Ⅱ期临床试验研究,如转移性黑色素瘤和软骨肉瘤[12-13]。既往研究发现[14],伊利司莫可以诱导细胞活性氧的生成、应激蛋白如热休克蛋白70(HSP70)的表达以及线粒体心磷脂的氧化,继而激活细胞色素C依赖的线粒体凋亡通路导致细胞死亡;另外,研究认为其抗肿瘤性也是通过调节氧化应激来实现的。然而,伊利司莫并未靶向线粒体并阻断呼吸链的电子转移[15],那么其到底通过何种途径产生活性氧呢?Wu等[16]采用液相色谱—质谱分析和单晶X射线衍射技术发现,伊利司莫与Cu2+可以按照1 ∶1(mol∶mol)的方式形成螯合物,且相较于Ni2+或者Fe2+,伊利司莫更容易与Cu2+形成稳定螯合物。随后研究发现,伊利司莫会优先结合细胞外Cu2+并以伊利司莫-Cu2+复合物形式,选择性地将Cu2+转运至肿瘤细胞的线粒体中[17]。当细胞外Cu2+缺乏时,伊利司莫则会失去其细胞毒性。当用不具备细胞膜渗透性的铜螯合剂浴铜灵螯合细胞外Cu2+后,伊利司莫对Cu2+的摄取和细胞毒性均被阻断[17]。伊利司莫-Cu2+复合物进入线粒体后,Cu2+被还原为Cu+,随即产生活性氧。伊利司莫-Cu2+复合物解离后,伊利司莫从细胞内流出至胞外,并重复形成新的伊利司莫-Cu2+复合物,将Cu2+从细胞外转运至细胞内,导致线粒体内铜的持续积累,最终诱导细胞凋亡[18]。值得注意的是,伊利司莫所表现出的线粒体选择性是该化合物一个独特特征,其他铜离子螯合剂(包括双硫仑)则不具有该特征[18]。
一项Ⅲ期临床研究的结果显示[19],在与紫杉醇联合应用治疗未加选择的晚期黑色素瘤患者时,伊利司莫并未诱导显著的临床反应。但如果依据黑色素瘤患者的血清乳酸脱氢酶(lactate dehydrogenase,LDH)水平对患者进行分组,相比高水平LDH患者,LDH水平较低或正常的患者在联合伊利司莫的方案中收获了较好的临床效果。LDH水平与肿瘤氧供需水平有关,高水平LDH意味着肿瘤细胞处于缺氧状态[20],由此提示伊利司莫靶向代谢活跃的线粒体并在氧合良好的细胞中发挥作用,也提示其可能依赖于氧化磷酸化而不是糖酵解过程而发挥抗肿瘤活性。相似的结果也在离体细胞实验中得到证实[21]。
Golub团队对比了生理和缺氧状态下的细胞对伊利司莫诱导的细胞毒性的敏感性差异,结果同样发现,在缺氧条件下生长的细胞对伊利司莫诱导的细胞死亡敏感性较低[5];同时研究结果还显示,伊利司莫在依赖线粒体呼吸的细胞中所产生的细胞毒性较其在依赖糖酵解的细胞中所产生的细胞毒性强近1 000倍,进一步强调了细胞呼吸在介导铜诱导的细胞死亡过程中发挥的作用。此外,Soma等[22]证实,伊利司莫可以通过增加线粒体内Cu2+含量和恢复细胞色素C氧化酶活性而挽救酵母细胞的呼吸缺陷;细胞色素C氧化酶是线粒体呼吸链的末端酶,铜是其重要辅助因子。另一项研究指出[23],在结肠癌细胞中,伊利司莫和铜联合处理导致铜滞留在线粒体内,引发活性氧累积,进而导致溶质载体家族7成员11(solute carrier family 7 member 11,SLC7A11)降解,而SLC7A11与铁死亡密切相关,且应用铁死亡抑制剂可以减轻伊利司莫诱导的细胞死亡。由此推测,铜诱导的细胞死亡可能是一种铜依赖的铁死亡方式。然而随后研究发现,与铁死亡抑制剂相比,线粒体抗氧化剂、脂肪酸或线粒体功能抑制剂更大程度地抑制了伊利司莫诱导的细胞死亡[5];同时,代谢组学分析结果显示,随着伊利司莫作用时间延长,非小细胞肺癌ABC1细胞所产生的三羧酸循环代谢产物明显增多[5];由此表明,Cu2+诱导的细胞死亡靶向的位置可能是三羧酸循环过程本身,而不是电子传递链过程,同样也并不是直接诱导铁死亡过程[5]。
全基因组CRISPR-Cas9功能缺失筛选确定了介导铜死亡的特定代谢途径。研究者采用了伊利司莫和DTC两种铜离子载体分别处理人卵巢癌细胞,从两者共同区间内确定10种可能与铜死亡相关的基因,其中包含7个正调控基因:铁氧还蛋白1(ferredoxin 1,FDX1)、硫辛酸合成酶(lipoic acid synthetase, LIAS)、脂酰转移酶1(lipoyltransferase, LIPT1)、二氢硫辛酰胺脱氢酶(dihydrolipoamide dehydrogenase, DLD)、二氢硫辛酰转乙酰基酶(dihydrolipoamide S-acetyltransferase, DLAT)、丙酮酸脱氢酶E1-α亚基(pyruvate dehydrogenase E1 alpha subunit, PDHA1)、丙酮酸脱氢酶E1-β亚基(pyruvate dehydrogenase E1 beta subunit, PDHB),以及3个负调控基因:金属调节转录因子1(metal regulatory transcription factor 1, MTF1)、谷氨酰胺酶(glutaminase, GLS)、细胞周期蛋白依赖性激酶抑制剂2A(cyclin-dependent kinase inhibitor 2A, CDKN2A)。既往研究已证实伊利司莫可以直接靶标FDX1基因,而FDX1编码一种还原酶,可将Cu2+还原成毒性更强的Cu+[9]。Zhang等[24]发现FDX1与ATP产生相关,并证实FDX1与葡萄糖代谢、脂肪酸氧化和氨基酸代谢密切相关[24]。与FDX1发挥的作用相类似,LIAS、LIPT1和DLD也参与了蛋白质脂酰化代谢的过程,而DLAT、PDHA1、PDHB、MTF1、GLS以及CDKN2A则与丙酮酸脱氢酶复合体的形成有关[5]。
由此确定,FDX1基因和蛋白质脂酰化过程在铜诱导细胞死亡中的关键调控作用。蛋白质脂酰化修饰是一种从细菌到哺乳动物均高度保守的赖氨酸翻译后修饰[25]。通过单个基因敲除研究进一步证实,FDX1和LIAS缺失致细胞产生针对铜诱导的细胞死亡的抵抗力,并通过数据库分析确认FDX1基因与蛋白质脂酰化间存在直接联系[5]。Cu2+与脂酰化的蛋白结合,导致DLAT脂酰化发生寡聚化,进而导致铁-硫簇蛋白的表达减少及铁-硫簇装配异常。铁-硫簇蛋白质在生命活动中发挥重要的功能, 包括电子链的传递、基因组稳定性的维持及基因表达的调控等[26]。
综上,铜死亡的机制可以简要表述为铜离子(包括Cu2+和Cu+)与线粒体呼吸过程中的三羧酸循环中的脂酰化成分直接结合,导致脂酰化蛋白质聚集,继而铁-硫簇蛋白表达下调,从而诱导蛋白质毒性应激并最终导致细胞死亡(图1)[5]。
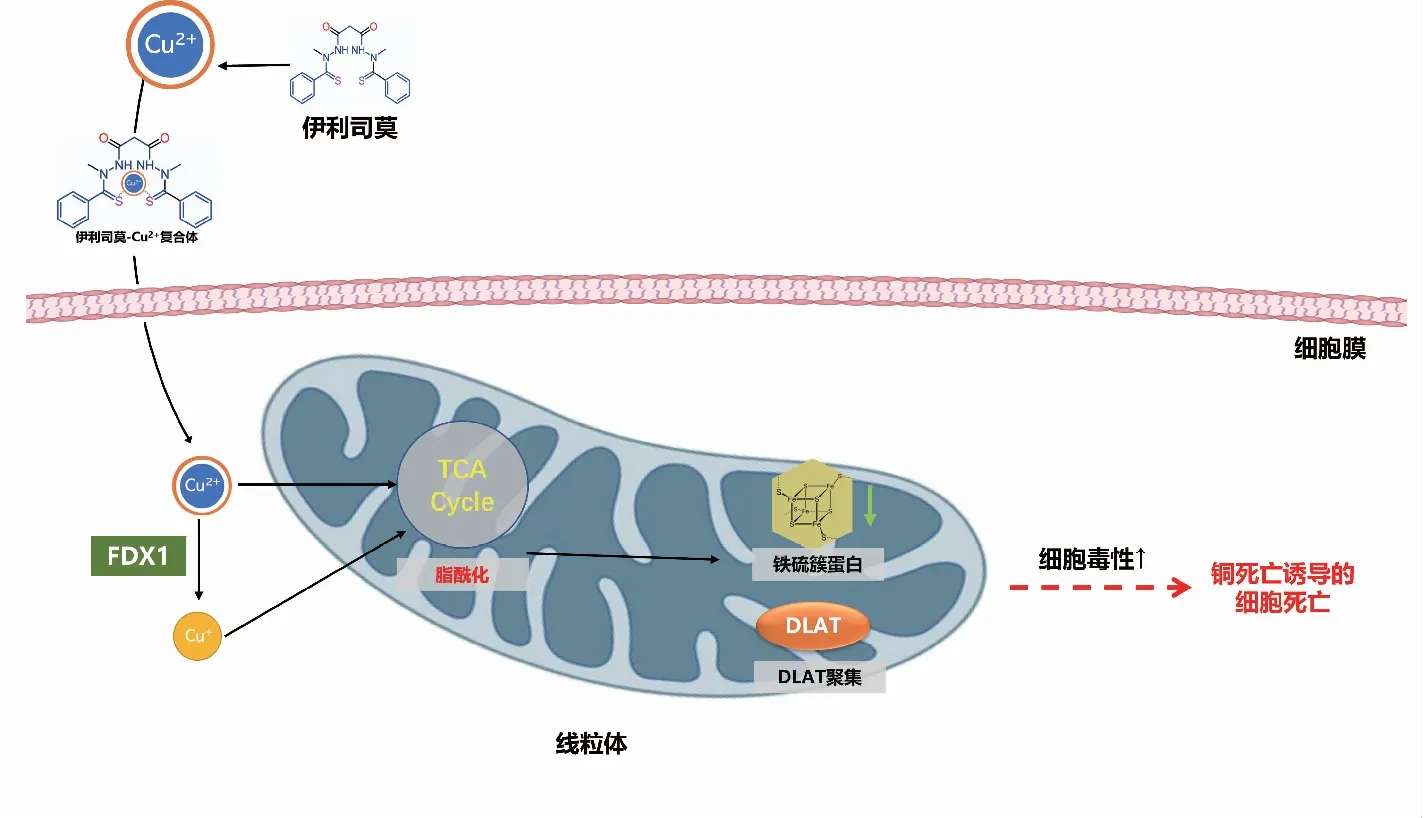
FDX1:铁氧还蛋白1;TCA cycle:三羧酸循环;DLAT:二氢硫辛酰转乙酰基酶
3 铜代谢相关疾病
铜在机体内的含量维持相对稳定,含量过低会破坏重要的金属结合酶功能,而过高则会导致细胞功能异常,引起细胞死亡[27]。机体内铜的摄入、排出以及代谢由多种因素共同调控并维持动态平衡,当体内铜稳态被打破,铜代谢异常或铜诱导的细胞死亡会导致一系列疾病的发生。
3.1 威尔逊病
威尔逊病(Wilson′s disease)于1912年被首次定义,是最经典的铜代谢异常疾病之一。根据铜在体内不同组织的蓄积,威尔逊病可表现为肝脏和神经功能障碍,包括肌张力障碍和帕金森病[28]。威尔逊病的致病基因定位于13q14.3,ATP7B基因的异常导致铜-ATP 酶功能受损、肝脏的铜解毒功能受损和体内铜超载[29],且ATP7B不同突变亚型导致了疾病的临床表现多样性[30]。Collins等[31]研究显示,针对ATP7B基因肽的直接检测越来越多地用于威尔逊病的临床诊断。此外,威尔逊病患者血清中铜同位素比率(65Cu/63Cu)变化也与健康人群不同,可作为评判疾病进展的指标,并且可以用来预测患者的预后情况[32]。威尔逊病的治疗可通过低铜饮食、螯合疗法或在极端情况下通过肝移植来实现,直接作用于肝细胞内的Cu+螯合剂或将成为威尔逊病的治疗方案[33]。在威尔逊病患者的驱铜治疗过程中,偶有铜缺乏情况的发生。据研究报道,威尔逊病患者铜缺乏时出现严重的周围神经损害及显著的中性粒细胞减少,而这些症状在血清铜水平恢复正常后得以改善,提示铜治疗方案也需要定期监测并维持铜平衡[34]。
3.2 Menkes病
Menkes病是一种罕见的X染色体隐性疾病,绝大多数患儿为男婴,但由于X染色体的随机失活,也有一些罕见女婴病例的报道[35]。既往报道Menkes发病率为1/298 000[36],但一项基于基因组聚合数据库估算的Menkes病发病率达1/ 8 664[37]。Menkes病的致病基因定位于Xq21.1,由ATP7A突变引起,与ATP7B导致铜蓄积相反,ATP7A突变导致的临床症状主要与铜缺乏相关[38]。CTR1可与ATP7A协同作用,调节细胞内铜离子水平和铜定向转运,例如,肠上皮细胞内层和脉络丛中,CTR1介导Cu+进入细胞内,而ATP7A促进铜从细胞中排出。Guthrie等[39]在动物实验中证实,基于伊利司莫-Cu2+复合物选择性释放线粒体Cu2+的机制,伊利司莫纠正了CTR1和ATP7A有缺陷的细胞膜铜离子转运能力,进而改善了Menkes小鼠的症状。
3.3 神经退行性疾病
在神经系统中,铜参与髓鞘形成,调节突触活动和信号级联反应以及调控神经元死亡。脑是仅次于肝脏的第二大含铜器官,在CTR1与 ATP7A转运下,铜离子可以透过血脑屏障分布至不同脑区。铜稳态失调已经在几种神经退行性疾病中得到证实,包括阿尔茨海默病(Alzheirmer′s disease, AD)[40]和帕金森病[41]。近来研究认为,血清中较高的游离铜水平与AD患者认知功能的衰退相关,其基本病理机制为铜离子过载引起β淀粉样蛋白(amyloid-β,Aβ)沉积与Tau蛋白异常折叠[42]。有研究表示,Cu2+不仅促进Aβ无定形聚集体的形成,还可增强对神经元的毒性,当其与Aβ结合后对神经元的毒性明显高于Aβ[43-44]。给予AD模型小鼠饮用水中添加铜可逆转低铜水平导致的Aβ产生和神经炎症的发生[45]。有研究证实,帕金森病患者血清和黑质中铜水平均较低[46]。α-突触核蛋白异常是帕金森病重要的病理特征,Cu2+、Cu+均可与其结合,而体外实验证实通过调控CTR1水平可以抑制 α-突触核蛋白折叠聚集,减轻帕金森病的病理改变[47]。此外,血清铜蓝蛋白水平降低可特征性地加重黑质铁沉积[48],而Ayton等[49]研究则显示,通过静脉注射铜蓝蛋白可能对帕金森病具有治疗有一定效果。
3.4 肥胖症
铜是脂肪分解的内源性调节器,其调控cAMP依赖的脂解作用以维持体重和能量储存的过程[50]。铜代谢异常可导致能量代谢紊乱,而铜依赖性酶的活性和丰度变化可能与肥胖症有关。研究发现,肝细胞中缺乏铜转运蛋白 ATP7B 的小鼠,其肝脏中出现铜积累并诱发肝脏脂肪变性和肥胖[51]。另有研究发现,ob/ob(瘦素突变体)小鼠和高果糖饮食大鼠肝组织中铜水平显著下降[52-53]。同样,非酒精性脂肪性肝病患者也会出现肝组织中铜水平下降[54]。但是,肥胖患者血清中铜水平升高,且与BMI和瘦素呈正相关,表明铜和(或)铜蓝蛋白可能与脂肪积累相关[55]。Bernier等[56]研究采用铜离子载体双硫仑治疗中年肥胖小鼠,结果显示其体重恢复正常且代谢功能障碍逆转。因此,纠正铜代谢紊乱有助于控制脂肪积累改善肥胖症状。
3.5 心血管疾病
既往研究指出,体内铜水平异常可能会诱发一系列心脏疾病,如缺血性心脏病[57]、心律失常[58]、心脏肥大[59]等,其机制可能是由于血清铜水平较低导致机体血脂代谢异常。最近研究发现,较高血清铜通过影响脂质代谢、低密度脂蛋白氧化及炎症反应加速动脉粥样斑块的形成,从而增加动脉粥样硬化性心脏病风险[60]。Cu2+可以通过氧化应激增加谷胱甘肽氧化,降低谷胱甘肽共轭程度,导致体内儿茶酚胺氧化继而对心脏产生毒性[61]。Li等[62]在动物实验中运用代谢组学分析,证实经铜暴露后猪心肌细胞中代谢物水平发生变化,其中7种上调,37种下调,其代谢物改变主要参与甘油磷脂代谢、脂肪酸的延伸与降解等过程,这些因素均与细胞自噬有关。研究显示,ATP7A在主动脉平滑肌细胞、主动脉内皮细胞中呈高表达,当ATP7A表达下调或功能障碍时则会通过上调 miR-125b促进主动脉瘤形成,miR-125b以铜依赖性方式增强促炎信号传导[63]。由此提示,血压调节、主动脉炎症或血管瘤可能与体内铜代谢及铜稳态有关。
3.6 肿瘤
多个研究证实铜代谢与肿瘤发生相关,与正常细胞相比,癌细胞对铜需求更高[64-67];部分癌症中表达大量脂酰化的线粒体蛋白,并表现出高强度的呼吸作用。多种癌症类型表现出瘤组织中铜金属含量的增高和(或)全身铜分布改变,包括乳腺癌[65]、宫颈癌[67]。值得注意的是,并不是所有肿瘤组织中铜离子水平均表现为异常升高,例如血液系统恶性肿瘤患者缓解期血清铜离子水平则较健康人明显降低[68]。Wang等[69]研究发现,阻断Cu2+运输,可引起细胞氧化应激并致细胞ATP水平降低,继而激活AMP活化蛋白激酶,从而导致脂肪生成减少,抑制肿瘤细胞的增殖。研究证实,铜与缺氧诱导因子-1α(hypoxia-inducible factor 1α,HIF-1α)表达水平密切相关[70-71]。HIF-1α可刺激血管生成,新生血管又可诱导血管内皮生长因子产生,继而导致肿瘤血管快速生成[72]。细胞运动介质1(mediator of ErbB2-driven cell motility 1, MEMO1)是一种铜依赖性氧化还原酶,进化相对保守,对细胞运动起重要作用。MacDonald等[73]研究发现,MEMO1是一种细胞内铜依赖性蛋白,其为体外乳腺癌细胞迁移、侵袭以及体内乳腺癌细胞自发性肺转移所必需。采用铜螯合剂四硫代钼酸盐可明显减少体内Cu2+含量,降低MEMO1表达,并显著减缓肿瘤的血管生成,抑制肿瘤生长,且对乳腺癌细胞侵袭能力有明显的影响[73]。Safi等[74]研究发现,在激素敏感和去势抵抗疾病模型中,双硫仑单独应用对前列腺癌细胞的生长无显著影响,而双硫仑与铜合用则明显地抑制前列腺癌细胞生长,其机制可能与前列腺癌细胞中强烈的CTR1依赖性铜摄取有关。在胃癌中也有类似发现,Wang等[75]采用双硫仑螯合Cu2+,通过Wnt/β-catenin通路调节应激,可抑制胃癌细胞生长。
3.7 新型冠状病毒肺炎
铜离子水平在新型冠状病毒肺炎(COVID-19)患者预后预测中具有一定价值。最近对武汉COVID-19患者全血微量元素的分析表明,病情较重患者血清铜含量普遍增加[76]。COVID-19患者疾病早期血清中Cu2+水平增高和Zn2+水平下降,且主要与炎症反应有关[77]。Hackler等[78]研究发现,COVID-19患者血清铜和硒水平有助于预测患者预后,在诊断为缺铜或缺硒的患者中补充含铜佐剂可能对疾病转归产生积极影响。另有研究指出,未能寻找到充分证据表明补充锌或铜可以预防COVID-19感染或成为重症病例[79- 80]。但是,仍有学者建议将铜作为佐剂或者采用纳米铜载体可能有助于COVID-19患者治疗[81-82]。
4 结语
综上所述,机体内的铜稳态通过调节铜的吸收、转运和排出来维持。研究发现,越来越多的疾病与铜代谢紊乱相关,特别是在肿瘤领域。铜死亡是一种新的细胞调节性死亡方式,其主要与三羧酸循环代谢紊乱有关;这种死亡方式与其他调节性死亡方式之间并不完全独立,其中可能存在紧密联系。铜死亡的发现有助于更深入地了解铜代谢疾病及其潜在分子机制,对于完善铜代谢及铜死亡机制探讨,筛选治疗铜代谢疾病的相关药物具有一定价值。
猜你喜欢
南京医科大学学报(自然科学版)(2022年8期)2022-11-22
中国交通信息化(2022年8期)2022-10-28
重庆理工大学学报(自然科学)(2022年9期)2022-10-26
中国种业(2022年9期)2022-10-13
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中学生数理化·高一版(2021年12期)2021-09-05
中国医药导报(2019年13期)2019-06-20
现代农业科技(2018年13期)2018-10-20
商界评论(2016年10期)2016-11-01
华人经济(2016年2期)2016-05-12
