
泉州湾宋代海船船体微生物群落结构多样性研究
2022-07-28 13:02:38费利华
文物保护与考古科学 2022年3期
费利华,覃 丹,唐 欢
[1. 福建省泉州海外交通史博物馆,福建泉州 362000;2. 馆藏文物有害生物控制研究国家文物局重点科研基地(重庆中国三峡博物馆),重庆 400015]
0 引 言
微生物是导致有机文物生物劣化的主要因素之一,同时也是文物保护领域长久以来面临的重大难题[1]。微生物种类繁多,代谢活动十分旺盛,代谢类型也呈现多样化,对木质文物破坏极大。木质文物的主要组成成分是碳、氢、氧和氮。构成木材的主要物质是纤维素和半纤维素(占70%)、木质素(占25%),其余5%为各种盐类、可溶性糖、酚和萜类等[2-3]。通常木材由管胞细胞组成,该细胞的细胞壁由许多细小的纤维丝组成,而在外层的初生壁中微纤维丝无规则的排列着,内层组成次生壁,因此纤维素是构成木材细胞壁的框架,是影响木材强度的主要因素。木质文物出土前多埋藏于土壤中或淹没在水体中,而土壤和水体均是微生物良好的栖息场所,具有微生物繁殖生长所需的各种因子(如pH、含水量、有机质等),为各类微生物的生长提供了有利条件[1,3-4]。由于水解作用,木材中的一些可溶性组分如单宁会溶解于水而流失,构成木材基本的成分多糖链或多肽部分或全部断裂,形成单链;由于微生物的作用,多糖链会被逐步分解成水和二氧化碳,并造成木质文物的腐烂。木质文物被腐蚀、降解后,化学成分与显微结构发生显著性的变化,许多较细的纤维组织消失了,木质的强度大大丧失[2-3,5]。微生物对木材的危害主要是木腐菌、着色菌和霉菌等真菌以及一些能够分解纤维素的细菌引起的各种损害。其中以木腐菌的破坏性最为严重,木腐菌通常是指能够侵蚀分解木材细胞壁而摄取其所需的养分致使木材腐朽的一类微生物,包括细菌和真菌,但主要以真菌为主[6]。着色菌与霉菌主要摄取木材细胞腔中的物质为养分,而对细胞壁物质无多大影响。着色菌仅仅改变木质的颜色,霉菌则主要引起文物生霉。生霉的主要影响是侵害木材、使木材局部酸性增加,滋生过程中留下各种色素污染木材等[6]。以微生物对木质文物中纤维素的分解为例,细菌中的好气性噬纤维菌(Sporocytophagasp.)、厌气性的梭菌(Clostridiumsp.)、维单胞菌属(Cellulomonassp.)、纤维弧菌属(Cellvibriosp.),真菌中的木霉属(Trichodermasp.)、曲霉属(Aspergillussp.)、青霉属(Penicilliumsp.)、蜡伞属(Hygrophorussp.)、奇果菌属(Grifolasp.),放线菌中的链霉菌等,均能分解纤维素,在纤维素酶的作用下,使木材中的全纤维素和木质素遭受到破坏,从而导致木质文物的损毁[7]。因此,木质文物中微生物类群的解析是众多木质文物预防性保护的重要前期基础,也是微生物病害发生时针对性高效防护的重要保障。
泉州湾宋代海船于1974年发掘出土,是我国发掘的第一艘体量大,年代早的远洋贸易木帆船,也是研究我国古代海外交通史、对外贸易史、造船史等不可多得的实物资料,属于国家珍贵一级文物,具有重要的保护价值[7-8]。泉州湾宋代海船的保护是国内乃至亚洲大型海洋出水木质文物保护的先例,出土后在当时有限的条件下采取先安装复原再缓慢自然阴干脱水的特有保护方式保存了船体,但未脱盐、复原安装时使用了大量的铁钉且长期处于一个相对开放的环境中展示[8-11]。这是泉州宋船特殊的保存状况。目前,船体结构基本稳定,但船体局部存在糟朽、表面降解等多种形式的病害。根据对泉州宋代海船保存现状的调查研究,船木中含有的盐分在开放式保存环境中产生的物理、化学作用是船体木材劣化的主要因素,但是否同时存在微生物作用值得探究,因此,极有必要对船木中存在的微生物类群开展研究。全面了解古船微生态现状,不仅有助于深入分析揭示船木劣化机理,同时有助于发现现存的微生物隐患问题,为后续微生物的预防性保护工作打下基础。因此,本研究通过对船体保存区域不同部位的木材进行了采样,采用高通量扩增子测序技术分析研究不同木材类型的组成的古船船体微生物的组成类群及分布规律,解析船体中是否存在对其本身有害的致腐微生物类群,为后续该船体的微生物病害防治工作提供重要的参考,也可以对泉州海外交通史博物馆目前对宋船的开放式展览的微生物病害风险进行评估,并为后续开展展览环境的整体调控提供参考依据。
1 材料与方法
1.1 古沉船样品
本实验所用样品采自福建省泉州市海外交通史博物馆保藏的宋代古沉船船体糟朽的不同部位,具体采样位置区分和木材材质详见表1。

表1 采集样本编号及所在古沉船部位
1.2 实验方法
1.2.1古船样本微生物的16SrRNA和ITS扩增子测序 用Mo Bio强力土壤DNA提取试剂盒提取采集到的古船木材样本总DNA。取5 μL样本DNA用1%琼脂糖凝胶电泳检测,超微量紫外分光光度计检测,使用无菌水稀释样本浓度至1 ng/μL,-20 ℃保存备用。以稀释后的基因组DNA为模板,设计并使用引物S-D-Bact-0341-b-S-17(5’-CCTACGGGNGGCWGCAG-3’)和S-D-Bact-0785-a-A-21(5’-GACTACHVG GGTATCTAATCC-3’)扩增细菌16SrRNA V3+V4高变区域[12],使用引物ITS3_KYO2(5’-GATGAAGAACGYAGYRAA-3’)和ITS4(5’-TCCTCCGCTTA TTGATATGC-3’)引物扩增真菌ITS2区域[13]。不同的样品的反向引物加上不同的标签序列以便于区分混合在一起测序的样品。所有的PCR反应体系均为30 μL,每个样本一式三份,PCR混合液包括:15 μL Phusion®高保真PCR Master Mix(New England Biolabs);0.5单位AccuPrimerTM Taq DNA聚合酶(Life Technologies,USA);0.2 μM正反向引物和10 ng模板DNA。PCR反应程序:98 ℃预变性1min,98 ℃变性10 s,50 ℃退火30 s,72 ℃延伸60 s,30个循环,最后72 ℃延伸5 min。待PCR反应结束后,取2 μL PCR产物经2%琼脂糖凝胶电泳检测。将三次PCR产物混合,经Gene JET凝胶提取试剂盒(Thermo Scientific)纯化,高灵敏度DNA芯片的Agilent 2100 Bioanalyzer(Agilent Technologies,Germany)分析纯化样品的粒径分布。再根据NEB Next®UltraTMDNA文库制备试剂盒的说明书制备文库并索引标记。Qubit@2.0 Fluorometer(Thermo Scientific,U.S.A)和Agilent Bioanalyzer 2100 system(Agilent Technologies,Germany)检测文库质量,最后由Illumina MiSeq测序平台上机测序(北京贝瑞和康生物技术有限公司,中国北京)。
1.2.2数据拼接和预处理 根据overlapping双末端序列FLASH软件(V1.2.7,http://ccb.jhu.edu/software/FLASH/)[3]合并正反引物序列,根据不同的标签序列将序列与其对应样品进行归类,获得raw tags。将raw tags经QIIME(V1.7.0,http://qiime.org/index.Html)软件[14]过滤掉连续高质量(≥Q20)碱基数小于总长度75%的tags,最终得到clean tags[15]检测获得的clean tags的质量。UCHIME算法(http://www.drive5.com/usearch/manual/uchime_algo.html)[16-17]检测并去除嵌合体,以便得到可用于下游分析用的有效tags。然后根据UPARSE软件包(Uparse v7.0.1001)使用UPARSE-OTU和UPARSE-OUT ref算法将得出的有效序列按照97%序列相似性聚类分成不同的可操作分类单元(Operational Taxonomic Units,OTUs),同时剔除所有样本范围内只出现一次的OTUs。最后用RDP classfier将每个OTU的代表序列进行物种注释,查阅词典将英文物种名称翻译成中文[18-19]。
1.3 统计分析
用QIIME软件包通过主成分分析(PCA)将获得的OTUs进行聚类分析。QIIME能够进行Weighted unifrac PCoA分析或者Unweighted unifrac PCoA分析,可以在OTU的水平上反映各样品间或者不同组间微生物群落结构的差异,即β多样性[20]。微生物类群间的系统发育关系用KRONA[21]进一步列出。计算α多样性指数Chao1,ACE,Shannon,Simpson和coverage,这些指数反映了不同样品微生物类群的多样性和丰度[22]。
2 结果与讨论
2.1 高通量测序数据统计及有效性分析
不同部位的古沉船样品的高通量扩增子测序得到的数据统计及质控信息如图1和图2所示。细菌测定结果表明,25个测序样本累计的原始序列经拼接和嵌合体过滤后得到3 004 779条平均长度为468 bp的细菌序列,在97%相似水平上将其归类得到2 500 948个OTUs(图1)。其中B19样品包含的细菌丰度最高,而B13样本包含的细菌丰度最低,各样品分布图如图1。真菌测定结果显示,25个测序样本累计的原始序列经拼接和嵌合体过滤后得到3 979 377条平均长度为303 bp的真菌序列,在97%相似水平上将其归类得到3 894 302个OTUs。其中B8样品包含的真菌丰度最高,而B23样本包含的真菌丰度最低,各样品分布图如图2。以上分析结果表明,本次所有待测样本中的细菌和真菌测序数据量及有效性均达到正常值,测序数据可以用于后续物种注释及多样性分析。

图1 宋船船体样品中细菌OTUs分布图Fig.1 Distribution of bacterial OTUs in different samples of the Song Dynasty shipwreck
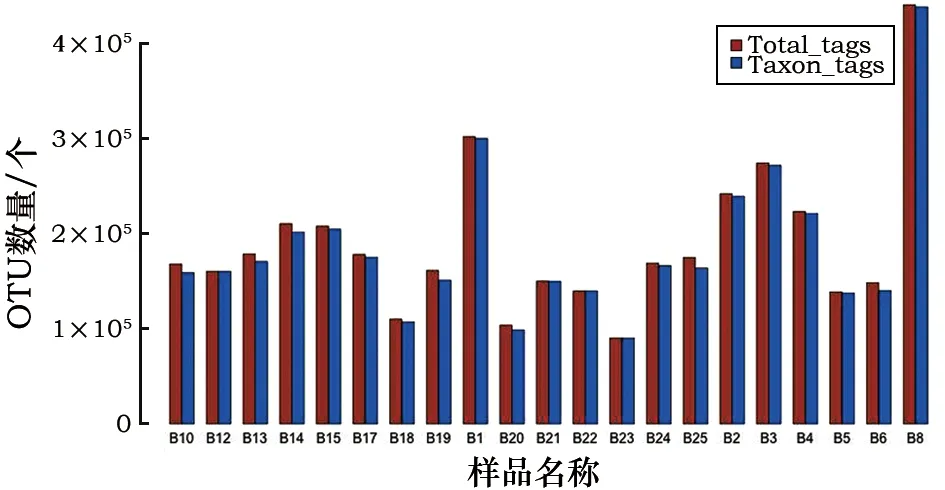
图2 宋船船体样品中真菌OTUs分布图Fig.2 Distribution of fungal OTUs in different samples of the Song Dynasty shipwreck
2.2 细菌多样性分析
为研究各样本的物种组成,对所有测序样本获得的Effective Tags,以0.97的一致性(Identity)进行OTUs聚类,然后对OTUs的序列进行物种注释[20]。根据每个OTU的细菌序列生成不同的细菌类群信息。分析结果表明,所有样品所含主要细菌类群在门分类水平上,变形菌门(Proteobacteria)为主要优势门类,占所有样品门类的78.08%,分布在所有样品中,其在样品B8(96.12%)中所占比例最高,样品B4(93.99%)、B2(93.21%)次之,在样品B21(4.00%)中所占比例最低;其次为厚壁菌门(Firmicutes)(13.23%),分布在所有样品中,其在样品B21(95.64%)中所占比例最高,样品B5(36.39%)、B17(25.40%)次之,在样品B8(0.42%)中所占比例最低;此外还有放线菌门(Actinobacteria)(5.72%),分布在所有样品中,其在样品B17(27.17%)中所占比例最高,样品B1(14.42%)、B10(10.74%)次之,在样品B21(0.16%)中所占比例最低;拟杆菌门(Bacteroidetes)(1.06%),分布在所有样品中,其在样品B22(36.70%)中所占比例最高,样品B24(26.25%)、B23(18.47%)次之,在样品B15(0.04%)中所占比例最低;此外还有一些丰度含量低于1%的细菌门类,分别是蓝藻菌门(Cyanobacteria)(0.20%)、酸杆菌门(Acidobacteria)(0.07%)、浮霉菌门(Planctomycetes)(0.07%)、绿弯菌门(Chloroflexi)(0.05%)、疣微菌门(Verrucomicrobia)(0.02%)以及未分类(Unclassfied)(0.23%)及其他门类(图3)。
在属分类水平上,所有样品所含主要细菌优势类群为不动杆菌属(Acinetobacter),占所有样品属类的13.65%,分布在所有样品中,其在样品B20(28.46%)中所占比例最高,样品B24(28.34%)、B4(24.85%)次之,在样品B21(1.10%)中所占比例最低;其次为芽孢杆菌属(Bacillus)(6.88%),分布在所有样品中,其在样品B21(69.06%)中所占比例最高,样品B5(18.26%)、B17(11.67%)次之,在样品B19(0.08%)中所占比例最低;假单胞菌属(Pseudomonas)(3.95%),分布在所有样品中,其在样品B3(14.73%)中所占比例最高,样品B15(11.79%)、B4(11.11%)次之,在样品B21(0.32%)中所占比例最低;马赛菌属(Massilia)(3.69%),分布在除样品B14以外的所有样品中,其在样品B23(13.23%)中所占比例最高,样品B20(11.76%)、B22(9.73%)次之,在样品B19(0.07%)中所占比例最低;短波单胞菌属(Brevundimonas)(3.61%),分布在所有样品中,其在样品B22(14.61%)中所占比例最高,样品B24(7.26%)、B6(6.52%)次之,在样品B13(0.18%)中所占比例最低;副球菌属(Paracoccus)(2.50%),分布在除样品B14以外的所有样品中,其在样品B22(12.34%)中所占比例最高,样品B6(5.58%)、B24(5.55%)次之,在样品B15(0.09%)中所占比例最低;甲基营养菌属(Methyloversatilis)(2.40%),分布在除样品B21和B24以外的所有样品中,其在样品B8(8.04%)中所占比例最高,样品B5(7.45%)、B23(6.33%)次之,在样品B22(0.02%)中所占比例最低;鞘氨醇单胞菌属(Sphingomonas)(2.36%),分布在除样品B14以外的所有样品中,其在样品B22(6.38%)中所占比例最高,样品B20(5.71%)、B24(5.30%)次之,在样品B1(0.17%)所占比例最低;其他属类有志贺氏菌属(Shigella)(1.92%),分布在除样品B19以外的所有样品中;链霉菌属(Streptomyces)(1.76%),分布在除样品B6、B8、B13和B15以外的所有样品中;弧菌属(Vbrio)(1.05%),分布在除样品B2、B19、B20和B24以外的所有样品中;甲基杆菌属(Methylobacterium)(1.07%),分布在除样品B15以外的所有样品中;耶尔森式菌属(Yersinia)(1.01%),分布在除样品B5、B13、B19和B24以外的所有样品中。除此之外,在所有样品中还存在许多未分类的(Unclassfied)(43.35%)菌属(图4),它们中的许多微生物只被鉴定到了门的分类水平,而属的分类水平暂时还不能够界定,由此说明目前的高通量测序技术方法还有待进一步改善和提升。以上分析结果表明,在门和属的分类水平上船体不同区域的真菌组成类群的种类高度相似,没有显著性差异,但不同样品间相同微生物类群的相对丰度含量存在一定的差异,且船体整体的真菌微生物类群保持了较好的稳定和多样性。

图3 宋船船体细菌在门类水平上相对丰度柱形图Fig.3 Column chart of relative abundance of bacteria in different samples at the phylum level

图4 宋船船体细菌在属的分类水平相对丰度柱形图Fig.4 Column chart of relative abundance of bacteria in different samples at the genus level
对所有样品高通量测序OTUs根据非加权unifrac两种距离对所有样品进行主坐标分析(Principal Coordinates Analysis,PCoA)(图5),以考察各样品物种的聚集情况。分析结果表明,样品B21与其他样品之间的矩阵距离整体相距均较远,表明样品B21与其他样品之间的细菌群落结构组成差别较大;对局部而言,样品B1、B13、B14和B15之间矩阵距离较近,说明这四个样品之间的细菌群落组成较为相似;同样地,样品B20、B22和B24,样品B18和B23,样品B3和B4之间的细菌群落结构也较为接近。以上分析结果表明,对于船体不同部位的细菌群落组成具有一定的差异性,但这种差异性与船体的木材类型和所处结构位置的关联性不明显。

图5 宋船船体不同样品的细菌类群PCoA分析Fig.5 PCoA analysis of bacterial groups in different samples of the Song Dynasty shipwreck
2.3 真菌多样性分析
根据每个OTU的真菌序列生成不同的真菌类群信息,所有样品所含主要真菌类群在门分类水平上,子囊菌门(Ascomycota)为主要优势门类,占所有样品门类的93.68%,分布于所有样品中,其在样品B23(99.86%)中所占比例最高,样品B8(99.67%)、B2(98.04%)次之,在样品B1(86.35%)中相对丰度含量最低;其次为担子菌门(Basidiomycota)(4.19%),分布在所有样品中,其在样品B12(13.12%)中相对丰度含量最高,样品B4(9.07%)、B19(6.03%)次之,在样品B23(<0.01%)中所占比例最低;还有球囊菌门(Glomeromycota)(<0.01%)仅在样品B25中有极少量分布,以及一些未分类(Unclassfied)(2.13%)门类(图6)。由此可见,在门的分类水平上船体不同区域的真菌群落组成高度相似。
在属分类水平上,所有样品所含主要真菌类群中枝孢霉属(Cladosporium)为主要优势属类,占所有样品属类的12.26%,分布于所有样品中,其中在样品B17(36.02%)中所占比例最高,样品B22(30.88%)次之,在样品B6(1.07%)中所占比例最低;其次为假丝酵母菌属(Candida)(5.93%),分布于除样品B14、B15、B20、B22和B24以外的所有样品中,其在样品B23(59.41%)中所占比例最高,样品B3(23.19%)次之,在样品B10(0.01%)中所占比例最低;扁孔腔菌属(Lophiostoma)(5.66%)分布于除样品B2、B3、B4、B8、B12、B21、B22和B23以外的所有样品中,其在样品B24(18.48%)中所占比例最高,样品B14(18.11%)次之,在样品B1(<0.01%)中分布极少;外瓶霉属(Hortaea)(3.15%)分布于除样品B12和B23以外的所有样品中,其在样品B24(9.16%)中所占比例最高,样品B10(7.07%)次之,在样品B4(<0.01%)中分布极少;其他还存在一些丰度相对较低的真菌类群,包括毛壳属(Chaetomium)(2.66%)、曲霉属(Aspergillus)(1.64%)、枝氯霉属(Ramichloridium)(1.41%)、假尾孢菌属(Pseudocercospora)(1.05%)、德巴利氏酵母属(Debaryomyces)(0.68%)、小光壳属(Leptosphaerulina)(0.61%)、帚霉属(Scopulariopsis)(0.46%)、节担菌属(Wallemia)(0.40%)、隔孢伏革菌属(Peniophora)(0.30%)、原毛平革菌属(Phanerochaete)(0.24%)、毛孢耳属(Trichosporon)(0.19%)、尾孢菌属(Cercospora)(0.16%)、孢子丝菌属(Sporothrix)(0.13%)、伪暗球壳菌属(Paraphaeosphaeria)(0.13%)和弯孢菌属(Curvularia)(0.11%)等。遗憾的是,在本次微生物高通量测序中各个样品中也还存在大量的未鉴定到属的分类水平和许多完全未被分类水平鉴定(Unclassfied)(52.13%)的类群(图7),造成这种现象的原因可能是由于当前真菌类扩增子测序和分子技术的限制,有待进一步提升。以上分析结果表明,在门和属的分类水平上船体不同区域的真菌组成类群的种类高度相似,没有显著性差异,但不同样品间相同微生物类群的相对丰度含量存在一定的差异,且船体整体的真菌微生物类群保持了较好的稳定和多样性。

图6 宋船船体真菌在门类水平上相对丰度柱形图Fig.6 Column chart of relative abundance of fungi in different samples at the phylum level

图7 宋船船体真菌在属的分类水平相对丰度柱形图Fig.7 Column chart of relative abundance of fungi in different samples at the genus level
对所有样品高通量测序OTUs根据非加权unifrac两种距离对所有样品进行主坐标分析(图8),以考察各样品物种的聚集情况。结果显示,样品B4与其他样品之间的微生物群落组成差别较大,样品B8、B12和B22之间的微生物群落组成较为接近,样品B2、B3和B23之间的微生物群落组成较为接近,样品B1、B17和B21之间的微生物群落组成较为接近,其余样品之间的微生物群落组成均在不同程度上接近。以上的这些分析结果与细菌群落的分布组成具有类似的规律,即对于船体不同部位的真菌群落组成具有一定的差异性,但这种差异性与船体的木材类型和所处结构位置的关联性不显著。

图8 宋船船体不同样品的真菌类群PCoA分析Fig.8 PCoA analysis of fungal groups in different samples of the Song Dynasty shipwreck
2.4 讨论
1) 宋船船体微生物的来源。微生物种类繁多,具有代谢能力强、代谢方式多样、易变异且繁殖速度快等特点,能够适应各种各样的生境,因此广泛分布于各种环境中,形成了独特的微生物生态系统。有研究表明,馆藏有机文物的微生物来源途径主要包括文物本体发掘前期自身所携带的内生微生物、馆藏环境所扩散的微生物以及养护文博人员日常失误接触所带来的微生物[1]。前期的研究中,本课题组采用经典的传统分离培养的研究方法对该船的展存环境微生物类群进行了研究,研究发现芽孢杆菌属(Bacillus)和枝孢霉属(Cladosporium)是环境中主要的优势微生物类群[10]。本次高通量测序结果显示在船体的各个部位发现了类似的优势微生物类群的存在,例如不动杆菌属(Acinetobacter)、芽孢杆菌属(Bacillus)和枝孢霉属(Cladosporium)等,表明这些类群的微生物可能来源于船体展存的环境。同时,在本次研究中也发现了一些前期空气环境中未发现的微生物类群存在于船体中,包括细菌中的假单胞菌属(Pseudomonas)、马赛菌属(Massilia)、波单胞菌属(Brevundimonas)、副球菌属(Paracoccus),甲基营养菌属(Methyloversatilis),以及真菌中的枝氯霉属(Ramichloridium)、假尾孢菌属(Pseudocercospora)、节担菌属(Wallemia)和尾孢菌属(Cercospora)等。相关文献表明这些微生物类群多次在水环境、土壤环境以及植物内生组织中被发现[23-27],但在常见的环境微生物的分析研究中却未见报道[28-31],因此推测这些微生物类群可能源自于该船体发掘的水环境中,属于文物馆藏前本体所携带,但这一推测结果仍待进一步研究证实。
2) 宋船船体微生物群落的特殊性。值得关注的是,本次在船体中发现的一些细菌和真菌具有能够耐受高盐环境的特性,例如细菌中的优势菌属假单胞菌属(Pseudomonas)[32]、芽孢杆菌属(Yersinia)[33],真菌中的枝孢霉属(Cladosporium)[34]、曲霉属(Aspergillus)[35]均可在高盐环境中生长。曹宏明等在红树林根际土壤中分离获得了5株假单胞菌属(Pseudomonasspp.)的细菌菌株,其均可耐受70 g/L的高盐环境[32]。丰开庆等从土壤中分离获得一株芽孢杆菌属的细菌菌株TYUT105,可耐受50 g/L的高盐环境[33]。因此,根据相关文献的报道,可推测宋船船体中这些耐盐的微生物类群的存在可能与该条船体前期修复保护时未脱盐处理有关,船体长期处于一定盐浓度状态下进而影响了其船体微生物类群组成的特异性。
3) 宋船船体微生物的潜在危害。微生物是造成木材降解糟朽的重要生物因素之一[36]。细菌对木材的腐朽作用发生在几乎缺氧的状况下,比如长期埋在土里、置于水中或长期处于高湿状态,而真菌对木材的破坏程度与速度又远远高于细菌,特别是木材腐朽真菌[37]。大多纤维素降解真菌在生长过程中能产生菌丝,菌丝具有很强的穿透能力,能穿透植物角质层的阻碍,紧紧依附和穿插在纤维物质上,增大降解酶与纤维物质的接触面积,从而加快降解速率[38]。目前大量文献已经报道可降解木质纤维素的细菌、真菌和放线菌200余种[39],其中最为突出的类群是青霉菌(Penicillium)[40]、曲霉菌(Aspergillus)[41]、芽孢杆菌(Bacillus)[42]和链霉菌(Streptomyces)[43]。景如贤等[42]从常年落叶土壤中筛选到菌株芽孢杆菌属(Bacillus)D2,该菌株以3%的接种比例、pH值为7.0、温度37 ℃和转速160 r/min,培养3 d后得到最高酶活,纤维素酶活为1.456 IU/mL。宁振兴等[44]筛选到的塔宾曲霉(Aspergillustubingensis)GYC501菌株,培养36 h后,纤维素酶活高达418.2 U/mL。李季蓉等发现在一定条件下枝孢霉属枝孢霉属的纤维素降解酶活力高达2.09 IU/mL[45]。陈岳等对“南海一号”船体发掘期间的微生物研究发现,镰刀菌属是其主要的致病菌属,该属微生物类群对木材中的纤维素和木质素等物质具有较大的破坏性[46]。宫秀杰等和李季蓉等关于微生物产生纤维素分解酶活力的研究表明芽孢杆菌属(Bacillus)、链霉菌属(Streptomyces)、曲霉属(Aspergillus)、枝孢霉属(Cladosporium)等菌株都是高产纤维素酶的菌株[46-49]。与这些已有的研究相比,在本研究中船体不同部位的优势微生物类群中同样发现了大量芽孢杆菌属、链霉菌属、曲霉属和枝孢霉属等类群微生物的存在,由于已有的研究报道表明它们具有显著的纤维素分解能力,在一定条件下可能会对船体木材造成一定的降解糟朽破坏,因此,在后续该船体的预防性保护中应该定期对船体进行一定的微生物防治维护,以益于船体的长期保存。
4) 宋船船体微生物的整体防治策略。温度和相对湿度是影响馆藏文物保存的两项重要环境因素,同时也是造成文物生物病害的2个主控环境因子。较高的温湿度是微生物大量繁殖的有利条件,当温度超过25 ℃,相对湿度高于65%时,木质文物就容易滋生微生物,造成霉菌大面积蔓延,致使文物表面和内部的微生物群落失衡,进而造成强烈的腐蚀和破坏[50-52]。因此,严格控制馆藏文物的环境对预防和减缓文物微生物病害的发生与损伤具有重要意义。通常开放式环境下不可控的相对湿度导致高湿气候环境下木材含水率提高,进而提升微生物对木材的降解能力。在本研究中发现,整个船体不同糟朽部位采集的相对丰度最高的真菌属均为枝孢霉属(Cladosporium),研究发现枝孢菌在潮湿环境中易于生长[10,53-55],因此在船体的后期预防性保护中一定要考虑船体本身和环境温湿度隐患,尤其在整个船体的展存环境中增大对湿度的调控,避免枝孢霉属类菌株的大量繁殖,从而保护船体免受微生物降解侵害。
3 结 论
本次对泉州宋船船体不同区域的微生物群落组成调查与分析结果表明,船体各个部位中存在稳定且丰富的微生物类群。尽管船体不同部位的细菌和真菌群落组成具有一定的差异性,但这种差异性与船体的木材类型和所处结构位置的关联性不显著。其中船体不同保存区域中存在的优势细菌种属为不动杆菌属(Acinetobacter)、芽孢杆菌属(Bacillus)和假单胞菌属(Pseudomonas),真菌以枝孢霉属(Cladosporium)、假丝酵母菌属(Candida)和扁孔腔菌属(Lophiostoma)为优势类群。此外本次检测分析发现的芽孢杆菌属、链球菌属、枝孢霉属和曲霉属均可降解木材纤维素,这些微生物在一定环境条件下生长爆发则会威胁船体的保存,具有一定的潜在隐患。因此,在后续该船体的保护中应该定期对船体进行微生物病害防治处理,严格控制船体的展存环境温湿度,以益于船体的长期保存。
猜你喜欢
建筑与预算(2024年2期)2024-03-22 06:51:36
大自然探索(2024年1期)2024-02-29 09:10:32
舰船科学技术(2022年20期)2022-11-28 08:19:52
军事文摘(2021年16期)2021-11-05 08:49:06
江苏农业科学(2019年5期)2019-09-02 14:01:46
广东农业科学(2017年5期)2017-08-29 10:37:31
焊接(2015年9期)2015-07-18 11:03:51
海军医学杂志(2015年2期)2015-02-27 13:47:42
应用海洋学学报(2014年1期)2014-11-22 07:17:44
中国舰船研究(2014年6期)2014-05-14 06:45:17
