
误诊为脑梗死的MELAS综合征1例报道并文献复习
2022-07-28 11:10彭珊彭乔君杨飞
河南医学研究 2022年13期
彭珊,彭乔君,杨飞
(1.川北医学院第二临床医学院/南充市中心医院 康复医学科,四川 南充 637000;2.川北医学院附属医院 神经内科,四川 南充 637000)
线粒体疾病是一组罕见的线粒体功能障碍性疾病,全球发病率约为0.02%,通常是由线粒体DNA或核DNA缺陷(突变或缺失)所致。细胞中的能量主要来源于线粒体,因此该类疾病的临床症状通常表现在能量需求高的组织中,包括骨骼肌、心肌、大脑及内分泌系统等。由于线粒体疾病通常表现为症状群形式,可将它们归类为几种综合征[1],线粒体脑肌病伴高乳酸血症和卒中样发作(mitochondrial encephalomyopathy with lacticacidemia and stroke-like episodes,MELAS)综合征为其中最为常见的综合征之一,然而由于其临床症状多样性容易被误诊。本研究报告1例误诊为脑梗死的MELAS综合征并结合文献复习,从而提高对MELAS综合征的认识。
1 病例简介
1.1 一般资料患者,男,29岁,因“左侧肢体无力10 h”于2020年10月6日到当地县医院住院。患者10 a前曾因“枕叶梗死”病史而致“偏盲”,经治疗后好转,未遗留后遗症;曾因右股骨头坏死行“右股骨头置换术”;平时体力劳动差,活动容易劳累;无结核、肝炎、心律失常、心脏病、高血压及偏头痛史,无糖尿病和脑梗死等家族史。入院时查体:心率76次·min-1,呼吸20次·min-1,体温36.4 ℃,血压 118/78 mmHg(1 mmHg=0.133 kPa),神志清楚,精神尚可,体型瘦小,未见皮疹,未触及浅表淋巴结;眼球可见各方向活动,无眼震,瞳孔等大等圆(直径为0.3 cm,对光反应灵敏);无表情肌瘫痪;双肺呼吸音清晰,心脏听诊无明显杂音;腹部平软,肝脾肋下未触及;左侧肢体肌力3级,右侧肢体肌力5级,肌张力正常,左侧病理征(+),右侧病理征(-)。入院诊断:(1)脑梗死(不明原因型);(2)痫性发作。
1.2 诊治过程患者在朋友家中聚餐,无明显诱因出现左侧肢体无力,表现为行走不稳,向左侧倾斜,自认为饮酒所致,未重视,休息几小时后,发现左侧肢体力量变差,遂到当地县医院就诊。在就诊途中出现意识丧失,伴有四肢抽搐,小便失禁,症状持续约2 min后,患者呼之能应,意识逐渐恢复,但仍有左侧肢体无力。患者在当地医院急诊就诊,头颅CT示“右侧颞顶叶低密度影,考虑脑梗死”。辅助检查:血清乳酸水平为3.06 mmol·L-1,肝功能、肾功能、血常规、甲状腺功能、心肌损伤、抗中性粒细胞胞浆抗体、抗核抗体、抗心磷脂抗体等均未见明显异常。结合患者起病急,病程短,头颅CT病灶已经显影,当地医院初步诊断为青年卒中,给予丁苯酞(每次0.2 g,每日3次)、阿司匹林(每次100 mg,每日1次)经验性治疗,患者病情好转,但未能明确卒中原因。患者于2020年10月10日为进一步明确原因到某医院神经内科就诊,(因患者股骨头置换术,不能完善磁共振检查)为患者复查头颅CT并进行头颅CT增强扫描:右侧颞叶片状低密度影(图1A);增强后右侧片状低密度影无明显增强,其内血管显影较好(图1B)。心脏彩超和动态心电图均未发现明显异常。结合患者体型矮小,有运动不耐受情况,无脑血管病危险因素,既往“脑梗死”病灶消失,偏盲病史,本次痫性发作等表现,怀疑患者存在线粒体疾病,因此建议患者将血液标本送第三方实验室进行线粒体基因检测。线粒体疾病基因检测回示:线粒体DNA A3243G突变(图2)。最终诊断为MELAS综合征,给予艾地苯醌(每次30 mg,每日3次)、维生素B1(每次30 mg,每日3次)、甲钴胺(每次0.5 mg,每日3次)等改善代谢治疗,并进行康复治疗。3个月后随访患者情况良好,无明显不适。怀疑患者是否存在家族遗传情况,对患者的父母及兄弟进行线粒体基因验证,结果均为正常。
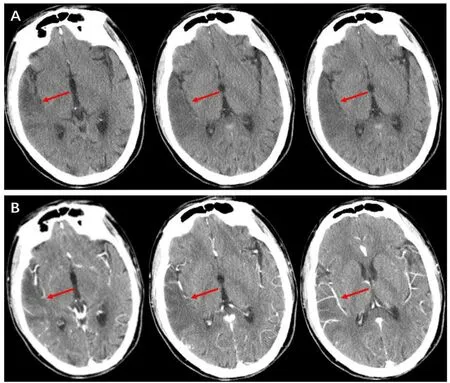
A为患者头颅CT平扫表现,右侧颞叶片状低密度影(红色箭头所示);B为患者头颅CT增强扫描表现,右侧片状低密度影无明显增强,其内血管显影较好(红色箭头所示)。

图2 线粒体疾病基因检测结果为线粒体DNA A3243G突变
2 讨论
2.1 临床特点MELAS综合征为母系遗传性罕见疾病,也存在部分散发病例,男性患者多于女性,男女发病的比例约为1.44∶1,多在2~31岁发病[2]。本研究中的患者无家族史,其父母及兄弟基因验证均为阴性,考虑为散发病例,患者发病年龄属于该病的流行病学高发期,且患者10 a前“枕叶梗死”导致偏盲,考虑为首次发作可能。MELAS综合征的临床症状和表现多样,有研究对MELAS综合征的临床特点进行分析,发现神经系统的症状主要有癫痫发作、偏瘫或偏身麻木、皮质盲或偏盲、运动不耐受、感音神经性耳聋、头痛、智能发育迟滞或痴呆等,非神经系统症状包括身材矮小、多毛、发热、呕吐、肾脏损害等[3]。本研究中患者的临床表现有偏瘫、癫痫发作、偏盲、运动不耐受及身材矮小等。实际上,MELAS常多种临床表现并发存在,这是导致其常被误诊的主要原因,因此需要注意识别。
2.2 辅助检查特征神经影像学检查和实验室生化检验可以为MELAS综合征的初步筛查提供帮助,病理活检也是支持诊断的重要依据[4]。然而,作为诊断MELAS综合征的金标准,基因检测的价值是无法替代的[5]。在生化检验方面,通常进行脑脊液和血液的乳酸水平检测,由于MELAS综合征是线粒体代谢功能障碍所致,从而造成无氧酵解功能增加,乳酸堆积,可能会在血液和脑脊液乳酸水平改变中发现线索。本研究中患者血乳酸水平也出现升高,但升高得并不显著。而且多种因素均可能影响到血乳酸水平,比如缺氧、运动、标本的收集等均可能导致血液乳酸水平上升,可能出现假阳性情况[6]。丙酮酸和乳酸的最小运动量试验也可以用于了解线粒体的代谢功能,但是,至今仍然无统一定量的运动标准。并且研究发现乳酸/丙酮酸比值只有升高到一定的程度才有意义[7]。
MELAS综合征的神经影像学特点为病灶并不根据血管供血区而分布,而且随时间的动态变化,病灶可能出现波动、迁移或消失[8]。病变多位于大脑皮质,常累及颞、顶、枕叶,急性期磁共振成像可见病灶表现为T1低信号,T2、FLAIR、DWI高信号[9]。本研究中患者由于股骨头置换,未能完成磁共振成像检查,但进行了头颅CT平扫及增强扫描。患者既往曾有“偏盲”病史且既往CT发现“枕叶病变”,而本次CT枕叶未见病变,出现颞叶病变,符合其影像结果随时间波动变化,而且在增强扫描中也发现患者血管充盈良好,并无明显异常。
MELAS的肌肉病理活检可作为对疑诊MELAS患者确诊的重要依据。肌活检冷冻切片经Gomori trichrome染色见不整红纤维,经糖原染色见糖原和脂肪堆积,组织内的血管壁经琥珀酸脱氢酶染色呈阳性等表现有助于诊断MELAS。但是上述病理表现并不是MELAS综合所特有,而且也并非能出现在MELAS患者的所有阶段,特别是发病早期[10],且其作为有创检查,患者接受度较低,本研究中患者不愿行肌肉活检。
线粒体DNA分析对诊断有决定性意义,目前检测线粒体DNA突变位点已成为诊断MELAS综合征的金标准[5]。 已有20多种线粒体DNA位点突变被发现与MELAS综合征的发病相关,如A3243G、T3271C、A3995G、G3959A、A3252G等,其中约80%的患者是由于线粒体DNA转运RNA亮氨酸基因位点3243的突变所致[10]。本研究中患者也为该点位的突变所致。因此,当怀疑存在MELAS综合征时,推荐进行基因检测。并且血液和尿液均可进行基因检测,这大大减轻了患者因担心检查损害而带来的阻力,既方便又准确。
2.3 治疗方法目前,国家药品监督管理局尚未批准治疗线粒体疾病的特效药物,临床以对症治疗为主,旨在减缓进展和缓解症状[11]。饮食上需减少内源性毒性代谢产物的产生,高蛋白、高碳水化合物、低脂饮食能够代偿受损的糖异生和减少脂肪的分解[12]。此外,减少使用线粒体毒性药物可延缓疾病进展,如本研究中患者存在痫性发作,就要避免选择丙戊酸、卡马西平、苯妥英和苯巴比妥等抗癫痫药物,其均伴有线粒体毒性[13]。而使用艾地苯醌、三磷酸腺苷、辅酶Q10以及大剂量B族维生素等可降低血乳酸和丙酮酸水平,左卡尼汀也可改善能量代谢以及促进脂类代谢。另外,应对伴发的癫痫发作、糖尿病、颅压增高及心脏病等疾病进行对症治疗。线粒体DNA突变的基因治疗研究还有待进一步探索,有望很快应用于临床治疗[14-15]。MELAS是一种高发病率和病死率的疾病。研究表明,中枢神经系统参与越早,MELAS可能越严重;患者的总体预后与发病年龄和临床症状密切相关;因而,早期发现并准确诊断和进行有效干预就显得尤为重要[16]。
线粒体疾病由于其遗传异质性和广泛的临床特征而具有挑战性。临床上MELAS综合征常被误诊为脑梗死、重症肌无力、肝豆状核变性等疾病[9]。因此,更详细地询问MELAS综合征患者的病史并进行相关辅助检查,以及对疾病轨迹的准确估计,有助于对MELAS患者进行诊断和治疗,降低MELAS的误诊率,改善患者的预后。本研究提示在诊断患者为青年卒中时,即使患者有急性卒中样临床表现,影像存在卒中样病灶,既往有可疑的脑梗死病史,但无脑血管病危险因素且病灶波动变化时,应考虑排除患者是否存在MELAS综合征。
猜你喜欢
医学研究生学报(2022年5期)2022-12-07
临床肺科杂志(2022年3期)2022-11-26
中华实用诊断与治疗杂志(2022年1期)2022-08-31
健康体检与管理(2022年4期)2022-05-13
现代临床医学(2022年2期)2022-04-19
现代临床医学(2022年1期)2022-02-12
中国卒中杂志(2021年7期)2021-11-29
家庭医学·下半月(2019年5期)2019-07-12
腹腔镜外科杂志(2016年12期)2016-06-01
恋爱婚姻家庭·养生版(2016年2期)2016-02-17
