
壳聚糖对四环素差向异构体的吸附差异研究
2022-07-21 07:15:26章耀鹏沈忱思
化工环保 2022年3期
张 冉,章耀鹏,陈 杨,沈忱思
(1. 东华大学 环境科学与工程学院,上海 201620;2. 宁波大学 科学技术学院,浙江 慈溪 315302)
近年来,四环素类抗生素的使用数量在全球范围内广泛增加,其中,四环素(TC)作为一种广谱抗生素,通常直接释放到水环境中,对人体健康和水生生态系统造成巨大威胁[1]。由于四环素类抗生素含有多个可离解的官能团,导致其化学性质不稳定,在水中极易发生异构化,生成四环类素差向异构体[2-5]。该类差向异构体的半衰期相对较长,细胞毒性比母体药物大70倍,已被欧盟确定为四环素、金霉素和土霉素的残留标识物[6-9]。因此,在预防与消除四环素类母体污染物的同时,其异构化产物急需关注。
去除水体中四环素类抗生素的方法主要有化学氧化法、电解法、光催化法、吸附法以及微生物法等[10-13]。其中,吸附法因其低成本、高效率、易操作等优点被广泛应用。然而,目前多数研究的重点都集中在母体污染物的消除上,关注异构体污染物的处理技术几乎没有[9,14,15]。四环类素差向异构体在吸附行为和活性特征上表现出与母体药物不同的性质。例如,HALLING-SØRENSEN等[5]研究发现四环类素差向异构体在土壤表面的吸附行为较母体药物减弱,增加了其向水体中迁移的能力,产生的环境生态风险不可忽视。这样的改变也极有可能影响四环素类抗生素在水处理过程中与处理材料或试剂间的相互作用,对处理效果产生影响。
壳聚糖作为一种具有双螺旋结构和氨基葡萄糖单元的天然高分子,因其优异的吸附性能和生物降解性而广受关注[16]。本研究以壳聚糖作为吸附剂,探究了其对TC和差向四环素(ETC)的吸附去除效果,并对吸附影响因素和吸附机理进行了研究和分析,为有效降低四环素类抗生素异构化带来的环境生态风险提供科学依据。
1 实验部分
1.1 试剂和仪器
壳聚糖、盐酸、柠檬酸(CA)、Na2HPO4、NaOH来自国药集团化学试剂有限公司,均为分析纯。TC、ETC、乙腈来自百灵威科技有限公司,甲酸来自上海安谱实验科技股份有限公司,其中TC和ETC纯度为90%~98%,乙腈和甲酸为色谱纯。
高效液相色谱/电喷雾电离-质谱联用仪(Waters 3100型,上海沃特世公司),傅里叶变换红外分光光度计(Nicolet6700型,中国赛默飞公司),X射线光电子能谱仪(ESCALAB 250XI型,中国赛默飞公司)、三维荧光光谱仪(Hitachi F-7000型,日本日立公司)。
1.2 吸附实验
称取一定量的壳聚糖加入到100 mL TC或ETC溶液中,通过改变单一因素(壳聚糖投加量、吸附pH、污染物初始质量浓度、单一/混合溶液)条件进行分批吸附实验,其中pH通过CA(0.01 mol/L)-Na2HPO4(0.02 mol/L)-NaOH(0.1 mol/L)缓冲体系调节。所有实验均在25 ℃搅拌条件下进行,每隔一段时间用1 mL注射器取样待测,每个条件下均设置3个平行实验。
1.3 分析方法
采用高效液相色谱/电喷雾电离-质谱联用仪测定TC/ETC的质量浓度:色谱柱为Waters Atlantis T3型,2.1 mm×150 mm×5 μm,柱温为30 ℃;流动相由1%(体积分数,下同)甲酸水溶液(80%)和1%甲酸乙腈溶液(20%)组成,流量为0.2 mL/min;进样体积为10 μL;紫外检测波长280 nm;毛细管电压3.0 kV,源温度120 ℃,去溶剂化温度400 ℃,去溶剂化气体流量400 L/h,倍增器电压750 V;全扫质荷比范围设置为200~800。
吸附剂的表征:采用FTIR在波数400~4 000 cm-1范围内分析吸附剂的官能团和物质结构;通过XPS分析吸附剂表面的元素化学状态;采用EEM在激发波长和发射波长分别为300~600 nm和350~650 nm范围内分析吸附剂的EEM光谱。
2 结果与讨论
2.1 壳聚糖对TC和ETC的吸附效果
在未调节吸附pH、污染物初始质量浓度为1×10-6mol/L的条件下,壳聚糖投加量对TC和ETC去除率的影响如图1所示。

图1 壳聚糖投加量对TC和ETC去除率的影响
随着壳聚糖的投加量从1 g/L增加到3 g/L,TC和ETC的去除率也逐渐增加。从长时间看,ETC的去除率略高于TC,3 g/L壳聚糖对TC和ETC的去除率在24 h后分别达到了69.75%和75.04%。但在短时间内,ETC的去除率明显高于TC,尤其是壳聚糖投加量较少时差距更为显著,1 g/L壳聚糖对TC及ETC的去除率在6 h时分别为31.74%和46.33%。
pH是影响四环素存在形态及吸附效率的重要因素。在壳聚糖投加量为3 g/L、污染物初始质量浓度为1×10-6mol/L的条件下,吸附pH对TC和ETC去除率的影响如图2所示。由图2可见:pH对TC和ETC吸附的影响趋势基本一致,在pH=6.03时吸附效果最佳;而随着pH的升高或降低,吸附效果均明显下降。虽然吸附24 h后壳聚糖对TC及ETC的去除效果接近,但短时间内,尤其是工况条件不佳时,ETC的吸附速率较快。例如:在pH=6.03时,反应24 h后,ETC的去除率为83.15%,TC的去除率为77.26%;而在pH=4.07时,反应4 h后,ETC的去除率为54.37%,TC的去除率仅为34.31%。

图2 吸附pH对TC和ETC去除率的影响
TC是两性化合物,具有可变的电离官能团(图3)。在酸性条件下(pH<3.30),TC主要以阳离子形式存在;在弱酸及中性条件下(pH 3.30~7.68),TC以阴阳离子共存的方式存在;在碱性条件下(pH>7.68),TC主要以阴离子形式存在[17]。同时,壳聚糖作为吸附剂对pH条件也非常敏感,静电作用力被认为是壳聚糖吸附的最主要的作用力,在酸性条件下壳聚糖表面的—NH2基团易被质子化,可通过静电作用力吸附阴离子类污染物,而碱性条件下壳聚糖仅能通过氢键作用力及疏水作用力等相对较弱的作用力对污染物进行吸附[18]。酸性环境及碱性环境均会对TC在壳聚糖表面的吸附产生负面影响。当pH为酸性时,壳聚糖结构中的—NH2可质子化为—NH3+,但TC同时也以部分阳离子的形式存在,吸附作用力较弱;当pH为碱性时,壳聚糖结构中的—NH2无法质子化,而此时TC以阴离子形式存在,也无法较好地与壳聚糖结合;当pH处于弱酸至中性条件时,壳聚糖表面的—NH2部分质子化形成带正电的—NH3+,TC以阴阳离子共存的方式存在,二者可通过静电作用相结合。因此,弱酸性条件下,壳聚糖对TC和ETC的吸附效果最佳。

图3 TC的分子结构
在壳聚糖投加量为3 g/L、吸附pH为6.03的条件下,污染物初始浓度对TC和ETC去除率的影响如图4所示。结果表明,壳聚糖对TC和ETC的吸附去除率与其初始浓度呈现负相关,并且在12 h后基本达到了吸附平衡。
2.2 吸附动力学及等温吸附模型
为了进一步考察壳聚糖的吸附机制,分别利用准一级动力学和准二级动力学方程(式(1)和式(2))对图4的实验数据进行拟合,结果如表1所示。准一级动力学方程的R2整体上高于准二级动力学,表明壳聚糖对TC和ETC的吸附更符合准一级动力学方程。

表1 壳聚糖对TC和ETC吸附的动力学拟合参数
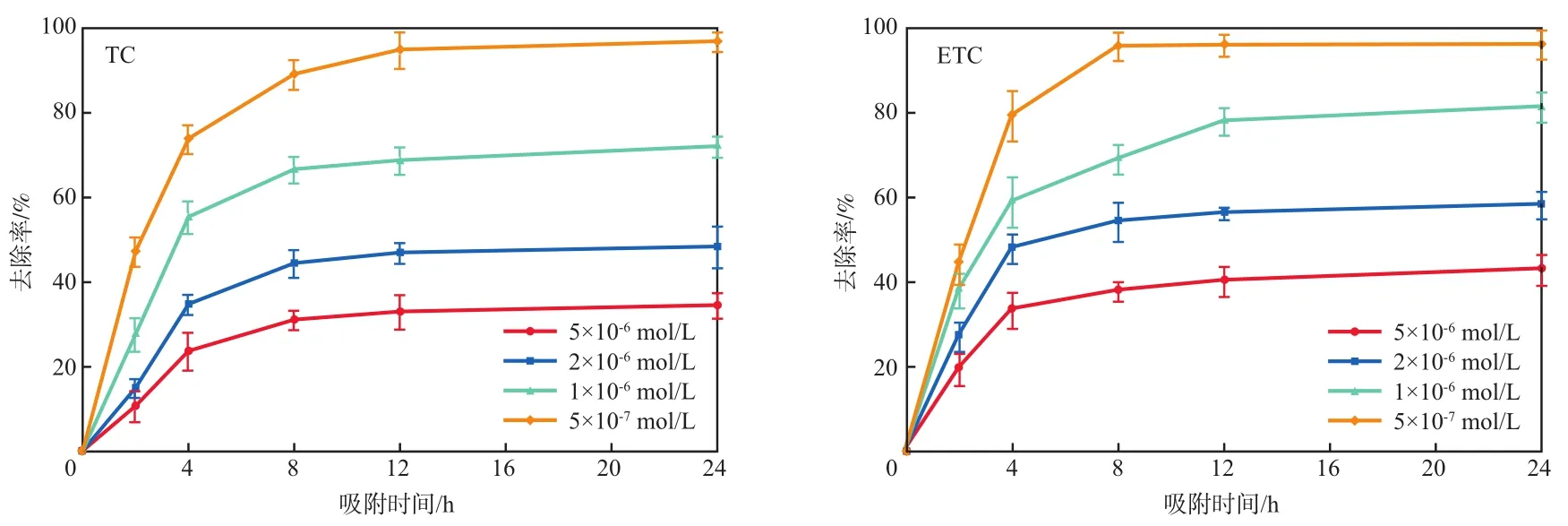
图4 污染物初始浓度对TC和ETC去除率的影响

式中:qe为平衡吸附量,mg/g;t为吸附时间,h;qt为t时刻的吸附量,mg/g;k1为准一级吸附速率常数,h-1;k2为准二级吸附速率常数,g/(mg·h)。
此外,本研究通过Langmuir和Freundlich等温吸附模型(式(3)和式(4))对吸附等温线实验(污染物初始浓度为0~5×10-4mol/L、壳聚糖投加量为3 g/L、吸附pH为6.03)数据进行拟合,结果如表2所示。从等温吸附模型的R2可以看出,Langmuir模型的R2更高,说明该吸附过程更符合Langmuir模型,更接近于单分子层吸附。
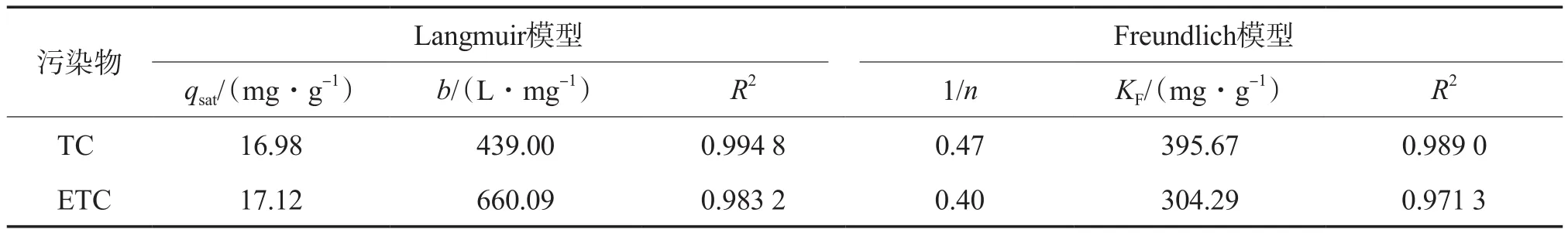
表2 等温吸附模型的拟合参数

式中:ρe为吸附平衡时的污染物质量浓度,mg/L;qe为平衡吸附量,mg/g;qsat为饱和吸附量,mg/g;b为Langmuir吸附平衡常数,L/mg;1/n为异质因子,与吸附效率相关;KF为Freundlich吸附平衡常数,mg/g。
2.3 机理分析
通过上述研究发现,在TC、ETC单独存在的体系中,不同吸附条件下壳聚糖对ETC的吸附去除效果均优于TC,尤其在高浓度、短时间的情况下,ETC的去除优势更为明显。在此基础上,本研究测试了TC和ETC共存条件下壳聚糖对TC及ETC的去除效果,结果如图5(TC和ETC初始浓度均为1×10-6mol/L、壳聚糖投加量为3 g/L、吸附pH为6.03)所示。相比污染物单独存在体系,共存体系中壳聚糖对TC及ETC的去除率有所下降,但ETC的去除效果依然显著优于TC,说明壳聚糖对TC和ETC的吸附性能具有一定的差异性。
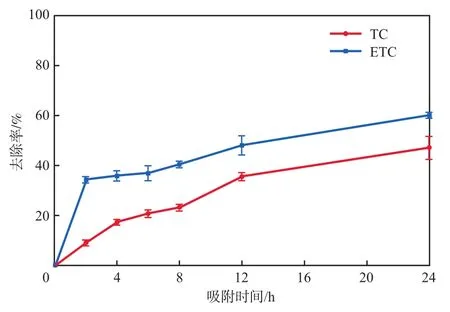
图5 壳聚糖对混合溶液中TC和ETC的吸附效果
图6为壳聚糖及其吸附TC(CS-TC)和ETC(CS-ETC)后的FTIR谱图。壳聚糖在3 430 cm-1处的吸收带是形成氢键缔合的—OH伸缩振动吸收峰与—NH的伸缩振动吸收峰重叠而增宽的多重吸收峰[19]。壳聚糖分子中存在着大量的分子内与分子间氢键,因峰较宽,壳聚糖吸附TC或ETC前后无明显差异。1 633 cm-1处的谱带可能是壳聚糖中C=O与O—H的氢键造成的,在吸附TC或ETC前后也未发生明显变化。1 420 cm-1和1 378 cm-1处的吸收峰分别对应—CH2的弯曲振动及—CH3的对称变形振动,壳聚糖吸附TC和ETC后代表—CH3对称变形振动的峰明显增强,且吸附ETC后的峰强度显著大于吸附TC后的,这可能归因于TC和ETC结构中的—CH3基团,且峰强与其吸附量变化一致。位于1 080 cm-1处的特征吸收峰为C3—OH(二级羟基)的特征峰,位于1 030 cm-1处的特征峰为C6—OH(一级羟基)的特征峰[20-23]。吸附TC或ETC后,壳聚糖结构中的C6—OH特征峰显著减弱,这说明壳聚糖对TC或ETC的吸附可能通过氢键作用发生在C6—OH的一级羟基上。
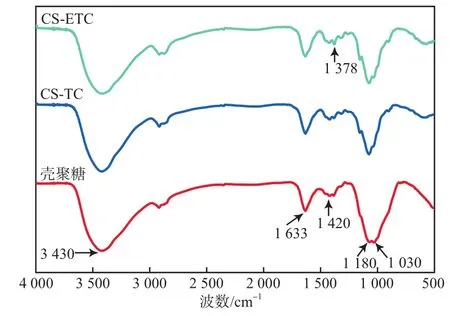
图6 壳聚糖吸附TC和ETC前后的FTIR谱图
图7为壳聚糖吸附TC和ETC前后N 1s的XPS谱图。位于结合能398.7 eV处的峰为—NH2的特征峰,399.5 eV处的为未脱乙酰化的乙酰氨基(—NHCO)特征峰,401.1 eV处的则归属于质子化的氨基(—NH3+)[24]。吸附TC或ETC后,壳聚糖结构中的—NH2及—NHCO的峰位均未发生明显变化,而—NH3+的特征峰明显向高结合能处位移,且峰面积变小,其占比从4.02%(壳聚糖)分别降至2.14%(CS-TC)和1.22%(CS-ETC),这说明壳聚糖的—NH3+在吸附TC和ETC的过程中发生了变化[25],可能通过静电作用力与TC和ETC结构中带负电的基团结合。相较之下,壳聚糖吸附ETC后的—NH3+峰面积变得更小,且结合能发生了更大的位移(401.4 eV)。这说明壳聚糖结构中的—NH2质子化后更易与ETC结合,使得壳聚糖对ETC具有更好的吸附能力。
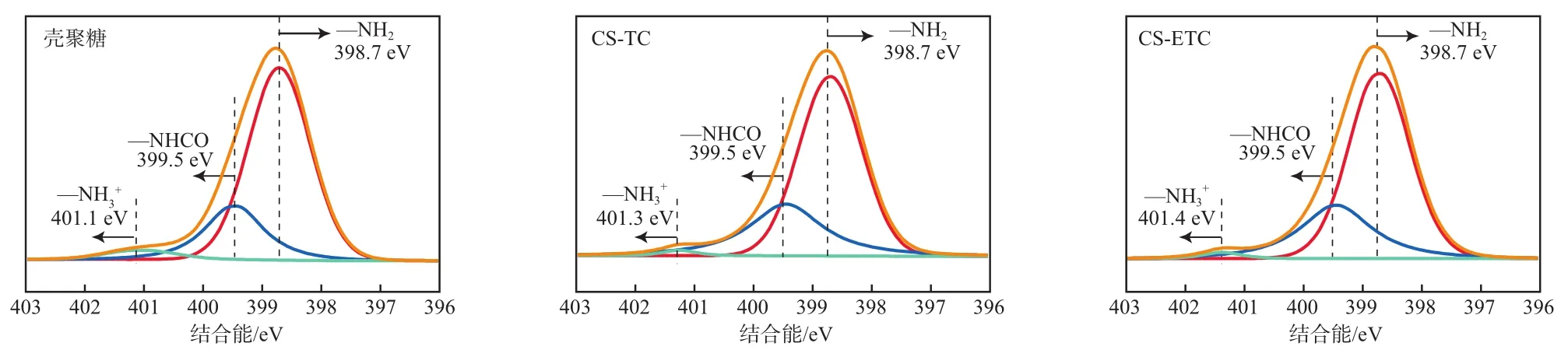
图7 壳聚糖吸附TC和ETC前后N 1s的XPS谱图
四环素类差向异构体与母体药物的差异源于C4位上的碳手性中心,差向异构化后C4-二甲氨基形成的空间位阻发生改变,使得异构化产物在吸附行为和活性特征上表现出与母体药物不同的性质[5,26]。壳聚糖对ETC的选择性吸附作用可能源于壳聚糖大分子的双螺旋结构及氨基葡萄糖单元,其螺旋沟槽能与有机小分子特异性络合,氨基葡萄糖单元又具有多手性中心,使得壳聚糖具有优异的立体选择性[20-23]。图8为污染物和吸附剂的EEM谱图。TC和ETC在激发波长(λex)/发射波长(λem)为(425~450)nm/(525~575)nm处出现一个主峰,且TC较ETC的荧光强度更大,说明TC在结构上刚性更强。壳聚糖在(329~348)nm/(379~418)处(A峰)和(369~378)nm/(423~453)处(B峰)含有两个自发荧光特征峰;当吸附TC和ETC后,特征峰的位置未发生明显位移,但荧光强度明显降低,说明TC和ETC的吸附改变了壳聚糖的立体结构从而对其自发荧光具有一定的猝灭作用。此外,壳聚糖与ETC结合后(CS-ETC)荧光的淬灭效应明显较低,两个特征峰均清晰可见,表明壳聚糖螺旋沟槽能与ETC较好地结合,其立体结构改变较少,这可能是壳聚糖对ETC具有更好吸附性能的主要原因。
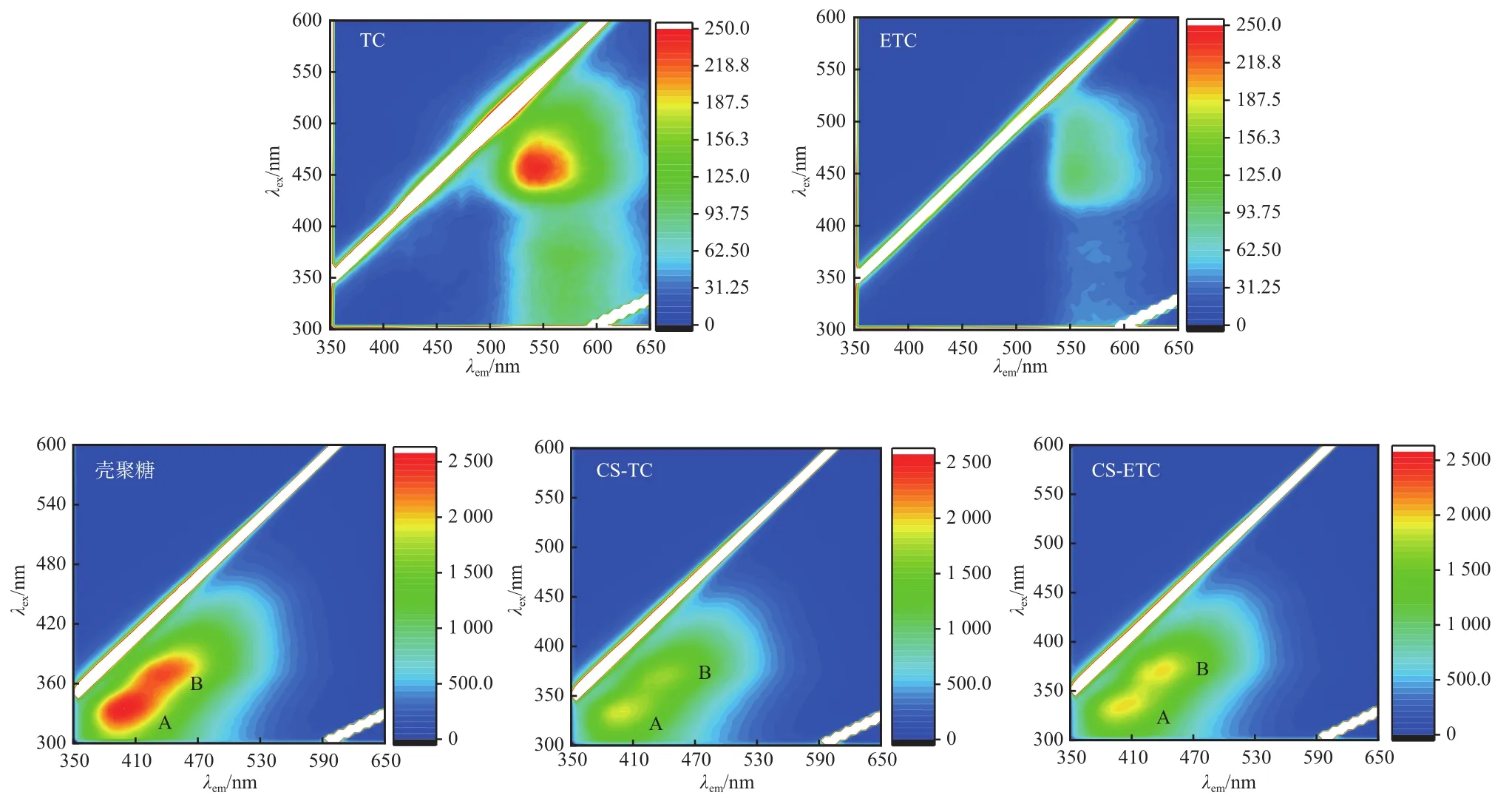
图8 污染物和吸附剂的EEM谱图
3 结论
a)不同吸附条件下,壳聚糖对ETC的吸附去除率均优于TC,尤其在短时间内,ETC的去除优势更为明显。吸附pH为6.03时,壳聚糖对TC和ETC的吸附去除效果最佳。壳聚糖对TC和ETC的吸附去除率与污染物初始浓度呈现负相关,随壳聚糖投加量的增大而增加。
b)吸附动力学拟合结果表明,准二级动力学方程更适用于壳聚糖对TC和ETC的吸附过程。等温吸附模型拟合结果显示,壳聚糖对TC和ETC的吸附更符合Langmuir模型。
c)FTIR、XPS和EEM表征结果表明,壳聚糖结构中的C6—OH和—NH3+可能通过氢键或静电作用力与TC和ETC结合,且壳聚糖的螺旋沟槽与ETC具有较强的结合能力,可能是壳聚糖对ETC具有更好吸附性能的主要原因。
猜你喜欢
工业安全与环保(2022年10期)2022-10-28 12:15:26
浙江大学学报(理学版)(2020年1期)2020-03-12 05:55:10
西安文理学院学报(自然科学版)(2016年4期)2016-12-19 08:18:59
中学生数理化·高三版(2016年2期)2016-09-10 07:22:44
China International Studies(2016年3期)2016-07-14 03:00:06
天津城建大学学报(2015年5期)2015-12-09 01:26:53
质谱学报(2015年5期)2015-03-01 03:18:25
安徽农学通报(2015年2期)2015-02-12 00:26:41
中国卫生(2014年5期)2014-11-10 02:11:32
机械与电子(2014年2期)2014-02-28 02:07:43
