
钾掺杂g-C3N4薄膜光阳极的制备及光电催化氧化降解水中双氯芬酸钠性能
2022-07-18 02:06:28龚妍熹王建兵柴歩瑜韩元春马云飞贾超敏
高等学校化学学报 2022年6期
龚妍熹, 王建兵, 柴歩瑜, 韩元春, 马云飞, 贾超敏
钾掺杂g-C3N4薄膜光阳极的制备及光电催化氧化降解水中双氯芬酸钠性能
龚妍熹, 王建兵, 柴歩瑜, 韩元春, 马云飞, 贾超敏
(中国矿业大学(北京)化学与环境工程学院, 北京 100083)
兼具高光学质量和电化学性能的薄膜光电极难以制备, 限制了光电催化氧化技术在水处理中的的应用. 本文采用原位煅烧法制备了负载在氧化铟锡(ITO)玻璃上的石墨相氮化碳(g-C3N4)薄膜电极, 并采用掺杂K+提高其光电催化氧化性能, 对电极进行了表征, 研究了其光电催化氧化降解水中双氯芬酸钠(DCF)的效率及降解路径. 结果表明, 原位煅烧法能制备出高质量的K+/g-C3N4薄膜光电极, K+的掺杂并未明显改变电极上g-C3N4的晶型、 价态和多孔形貌, 但可以提高ITO玻璃上g-C3N4的负载量, 增强电极对可见光的响应; K+的最佳掺杂浓度为0.002 mol/L, K+/g-C3N4薄膜电极光电催化氧化降解DCF的速率常数是纯g-C3N4薄膜电极的1.86倍; 当初始pH为4, 电压为1 V, 光源强度为0.96 W/cm2, 反应2 h后水中DCF降解率达到70%. K+/g-C3N4薄膜电极光电催化氧化过程中, 光催化氧化和电化学氧化之间存在协同作用, 两者相互增强, 并提高了反应过程中光生 空穴(h+)和羟基自由基(·OH)浓度, 在这两种活性物质作用下, 水中DCF分别被h+氧化生成咔唑衍生物、 与·OH发生加成反应生成多羟基芳香化合物, 最后开环生成小分子物质.
钾掺杂石墨相氮化碳薄膜光阳极; 光电催化氧化; 双氯芬酸钠; 光催化氧化; 电化学氧化
近年来, 全球水环境中广泛存在的药物及其生态风险备受关注. 城市污水处理厂二级生化出水是药物排放到自然水体的最重要途径, 有必要对其进行深度处理, 但常规的混凝沉淀-过滤工艺对水中药物的去除率不高. 现有的研究表明, 高级氧化技术能有效地去除水中的药物. 近几十年来, 在众多的高级氧化技术中研究最多的是光催化氧化技术[1]. 光催化氧化技术多采用TiO2作为光催化剂. TiO2因具有无毒无害、 稳定性高、 成本低等优点而受到青睐[2,3], 但其禁带较宽, 对可见光的响应低, 导致其实际应用受到限制[4].
近年来, 具有可见光催化活性的石墨相氮化碳(g-C3N4)被认为是一种非常有潜力的光催化剂[5~7]. g-C3N4的禁带宽度为2.7 eV, 作为光催化剂具有制备方法简单、 成本低廉及环境友好等优点, 因而被广泛地研究. 现有的研究采用掺杂[8]、 金属复合[9]、 大分子组装[10]、 模板剂[11]和表面异质结设计[12]等多种方法提高g-C3N4的光催化活性. 为了便于作为水处理的光催化剂, g-C3N4被负载在基材上制成薄膜电极. 最新的研究表明, 采用涂布法将g-C3N4负载在氧化铟锡导电玻璃上, 能够实现光催化氧化和电化学氧化的协同, 明显提高水中酚的降解和矿化效率[13]. 然而g-C3N4的颗粒较大、 水溶性差, 采用常规涂布和印刷法制备的薄膜电极中, g-C3N4和ITO玻璃结合力弱, 因而负载量较低, 导电性和光催化活性仍需要大幅提高.
为了制备高光电质量的g-C3N4薄膜, 本文采用原位煅烧法制备了掺杂K+的g-C3N4薄膜电极, 对其进行了结构表征, 研究了其作为催化剂, 光电催化氧化降解水中双氯芬酸钠的性能, 还对DCF降解过程中的中间产物进行了检测分析, 进一步探索了光电催化的降解机理, 为促进g-C3N4作为水处理光催化剂的环境应用提供了有益补充.
1 实验部分
1.1 试剂与仪器
三聚氰酸(C3H3N3O3)和苯并胍胺(C9H9N5)均为分析纯, 购于日本东京化工厂; 双氯芬酸钠(DCF), 购于北京蓝弋化工产品有限责任公司, 氧化铟锡(ITO)导电玻璃(长10 cm, 宽2.5 cm)由深圳晶伟特公司加工. 所有溶液均由超纯水配制.
X’Pert PRO MPD型X射线衍射仪(XRD, 荷兰帕纳科公司); H-7500型透射电子显微镜(TEM)、 Hitachi S450型扫描电子显微镜(SEM)和U-3010型紫外-可见漫反射光谱仪(UV⁃Vis DRS, 日本日立公司); Perkin Elmer System 2000型傅里叶变换红外光谱仪(FTIR, 美国铂金埃尔默公司); ESCALAB 250Xi型X射线光电子能谱仪(XPS, 美国Thermo公司); CHI660D型电化学工作站(上海辰华公司); PLS-SXE300型氙灯光源(北京泊菲来公司).
1.2 实验过程
采用原位煅烧法制备薄膜电极. 将三聚氰酸和苯并胍胺按摩尔比1∶1与40 mL超纯水混合, 搅拌 6 h, 在7500 r/min条件下离心15 min, 去除2 mL上层清液; 将得到的牛奶状混合物直接涂覆在ITO导电玻璃上, 然后冷冻(温度-55 ℃)干燥48 h, 再放入石英舟中; 然后用锡箔纸将石英舟密封, 并置于管式炉中煅烧(管式炉为氮气气氛), 以升温速率3 ℃/min升温至500 ℃后, 保温3 h, 自然冷却后置于水中超声, 以去除表面附着不牢固的粉末后, 得到薄膜电极, 记为I/g-C3N4.
采用原位煅烧法制备K+掺杂的g-C3N4薄膜电极, 将三聚氰酸和苯并胍胺按摩尔比1∶1, 分别与 40 mL不同浓度(0.001, 0.002, 0.005, 0.01 mol/L)的氢氧化钾溶液混合, 其它步骤同上所述, 得到的薄膜电极标记为K+/g-C3N4.
参考文献[13]报道的涂布法制备薄膜电极. 将三聚氰胺于550 ℃下煅烧4 h, 制备g-C3N4粉末, 取2 g g-C3N4粉末与100 mL浓硫酸混合, 室温下搅拌8 h后, 倒入500 mL的超纯水中, 超声处理成片状, 然后离心分离去除未剥落的g-C3N4, 清洗除去残留的酸, 然后于80 ℃干燥过夜获得g-C3N4薄膜粉末. 将100 mg薄膜粉末分散在100 mL水中, 超声波处理6 h, 将ITO玻璃重复涂布3次, 每次涂布后于80 ℃干燥30 min, 最后超声去除表面附着不牢固的粉末后, 得到D/g-C3N4薄膜电极.
降解实验在CHI660D型电化学工作站上进行, 反应体系为三电极系统, 采用自制的石英玻璃电解池(5 cm×5 cm×6 cm), 对电极为铂丝(长70 mm, 直径0.2 mm), 参比电极为饱和甘汞电极, 工作电极为K+/g-C3N4薄膜电极(有效面积为15 cm2). 氙灯光源功率为500 W, 安装有400 nm的滤光片, 使氙灯发出可见光. 将反应器放在磁力搅拌器上, 使溶液处于均匀搅拌状态. 光电性能测试时, 使用的电解质溶液为Na2SO4水溶液, 浓度为0.05 mol/L. 降解反应过程中, DCF反应液体积为100 mL, 初始浓度为 10 mg/L.
1.3 分析方法
通过LC-20A型高效液相色谱仪(日本岛津公司)测定DCF浓度, 色谱柱为C18柱, 以醋酸钠缓冲液-甲醇(体积比40∶60)为流动相, 检测波长为197 nm, 柱温为35℃, 进样量为20 μL, 流速为 1 mL/min.
火力发电也是耗水大户,重点抓了阳城国际发电有限责任公司节水改造项目,采用国际上先进的高压变频供水系统后,年可节水700万m3。二期工程建成了干式除灰除渣系统和污水处理回用系统,年可节水740万m3。
的中间产物使用LCMS 8040型高效液相质谱联用仪(日本岛津公司)检测, EIS为离子源, 液相色谱柱是C8(S/N=60423719). 流动相为0.1%(体积分数)甲酸溶液(A)-甲醇(B), 以体积比2∶8等度洗脱, 流速为0.4 mL/min, 柱温为30 ℃, 正负离子扫描, 扫描范围是100~700/. 利用产物离子扫描模式(PIS)对检测到的中间产物进行结构分析, 电压采用25 V, 扫描范围为50~600/.
2 结果与讨论
2.1 K+/g-C3N4薄膜电极的表征
图1(A)~(D)显示了原始的ITO玻璃、 涂布法制备的g-C3N4薄膜电极、 原位煅烧法制备的g-C3N4薄膜电极和K+/g-C3N4薄膜电极的照片. 可见, 3种方法制备的膜电极上g-C3N4颗粒分布都较为均匀, 涂布法制备的膜电极上面g-C3N4颗粒较少, 原位煅烧法制备的膜电极上面g-C3N4颗粒较多, 这主要是由于涂布法制备膜电极时没有采用高温焙烧工序, 而原位煅烧法采用了这一步骤. 高温焙烧是增加g-C3N4颗粒与ITO玻璃之间黏合力的关键步骤. 如果不采用该步骤, 很多与ITO玻璃结合不紧密的g-C3N4颗粒会被超声去除掉, 或是在光电催化氧化实验中由于表面作用力的破坏而脱落. 在原位煅烧法中, 三聚氰酸和苯并胍胺在水溶液中混合得到牛奶状混合物, 将此牛奶状的混合物直接负载在ITO玻璃上, 由于表面的润湿性, 湿的混合物颗粒可以黏附在ITO玻璃上, 在后续的干燥和焙烧中, g-C3N4颗粒就能够直接在ITO玻璃上生长. 文献[14]采用了湿式生长法制备了g-C3N4负载量较大的薄膜电极, 是将牛奶状的混合物干燥获得颗粒后, 再负载在ITO玻璃上, 两种方法在本质上无区别, 但是采用干燥后的颗粒负载, 很难将颗粒均匀地铺在ITO玻璃上, 导致制备的玻璃电极上g-C3N4颗粒分布有时不均匀, 因而本文采用将牛奶状的混合物直接负载至ITO玻璃上.

Fig.1 Photographs of ITO glass (A), D/g⁃C3N4 film electrode(B), I/g⁃C3N4 film electrode(C), and K+/g⁃C3N4 film electrode(D)
为了进一步提高g-C3N4的光催化性能, 在制备g-C3N4前驱体时掺杂了不同浓度的K+, 获得了不同浓度的K+/g-C3N4薄膜电极. 图2示出了原位煅烧法制备的K+掺杂浓度分别为0, 0.001, 0.002, 0.005, 0.01 mol/L的薄膜电极的SEM照片. 可以看出, K+掺杂对膜电极的表面形貌影响较小, 电极均呈现出多孔结构. 对比I/g-C3N4薄膜电极和K+/g-C3N4薄膜电极的照片, 可见, K+/g-C3N4薄膜电极上的g-C3N4颗粒负载量比g-C3N4薄膜电极略微多一些[图1(C)和(D)]. 电极截面SEM照片[图3(A)和(B)]显示, ITO玻璃表面的K+/g-C3N4薄膜厚度为2.02 μm, 而g-C3N4薄膜厚度为1.70 μm, 由此可知, K+掺杂确实增加了g-C3N4在ITO玻璃上的负载量, 从而增加了电极表面的活性位点, K+/g-C3N4薄膜电极的光催化活性得到了提高. 文献[15]报道, 分别采用钠钙玻璃、 石英玻璃和ITO玻璃作为基底, 结果显示, 钠钙玻璃形成的g-CN薄膜最厚, 外观也更均匀透明. 说明钠钙玻璃更容易促进g-CN薄膜的形成, 煅烧过程中 g-CN的部分基团可以与钠钙玻璃中的Na+或Ca2+结合, 促进了g-CN在基底上的气相沉积. 因此, 推测K+可能起到了与Na+和Ca2+同样的作用, 在原位煅烧过程中, K+促进了g-C3N4在ITO玻璃上的生长, 形成了较厚但均匀透明的K+/g-C3N4薄膜.

Fig.2 SEM images of the K+/g⁃C3N4 thin film electrode with the K+ doping concentration of 0(A, A′), 0.001(B, B′), 0.002(C, C′), 0.005(D, D′), and 0.01 mol/L(E, E′)

Fig.3 SEM images of the cross section of K+/g⁃C3N4 film electrode(A) and I/g⁃C3N4 film electrode(B)
图4(A)为不同K+掺杂浓度的g-C3N4薄膜电极的XRD谱图. 可见, 所有的样品均有两个明显的衍射峰, 分别在2=13.8°, 27.4°. 在2=27.4°处较强峰是层状堆积结构的特征峰, 对应的晶面为(002). 在2=13.8°处的峰为均三嗪单元的面内结构峰, 对应的晶面为(100). 所有样品的XRD谱图中均没有检测到钾的特征峰, 有可能是其含量太低或者是没有形成晶体. 不同K+掺杂浓度的g-C3N4特征峰峰强度不同, 说明K+的掺杂能够影响g-C3N4晶体成型.
图4(B)是K+/g-C3N4薄膜电极的TEM照片, g-C3N4为层状结构的大块状颗粒、 颗粒厚度小, 透明可见, 粒径在50~500 nm之间不等. 颗粒边缘区域颜色很深, 明显为多层叠加的形式. 这是因为在样品制备过程中, 由于超声分散不均匀, 出现大量的片层叠加, 颜色加深.

Fig.4 XRD patterns of g⁃C3N4 thin film electrode doped with the different concentrations of K+(A) and TEM image of K+/g⁃C3N4 thin film electrode (B)
(A). g⁃C3N4; K+concentration/(mol·L-1);. 0.01;. 0.005;. 0.002;. 0.001.
由图5(A)可见, K+/g-C3N4在807 cm-1处的谱峰对应三嗪结构的简正振动, 1200~1650 cm-1范围内的若干峰对应的是C—N杂环的典型伸缩振动, 2900~3200 cm-1范围内的宽峰对应的是N—H3和O—H的伸缩振动, 说明合成的g-C3N4中有部分残留的氨基, 表面吸附有少量的H2O分子. 由图5(B)可见, K+/g-C3N4薄膜电极对可见光的响应迅速, 在短时间内就可以达到稳定状态. 当K+浓度为0.002 mol/L时, 光电流最大, 达到0.12 mA, 是纯g-C3N4的2.22倍. 当K+浓度为0.01 mol/L时, 光电流最小, 只有62 μA, 是纯g-C3N4的1.15倍. 说明K+的掺杂对电极改性具有良好的效果, 可以有效提高g-C3N4光生电子和空穴的分离效率以及迁移速率. 最佳掺杂浓度是0.002 mol/L, 因此, 后续降解实验均采用K+浓度为0.002 mol/L的K+/g-C3N4薄膜电极. Tauc plot法[16]拟合的g-C3N4禁带宽度数据[图5(C)和(D)]显示, K+浓度为0.001 mol/L时, K+/g-C3N4的禁带宽度(2.55 eV)最小; 随着掺杂的K+浓度增加, K+/g-C3N4的禁带宽度逐渐增大, 但也比纯g-C3N4的禁带宽度(2.72 eV)小. 说明掺杂K+可以降低g-C3N4的禁带宽度, 从而提高了g-C3N4的光吸收能力, 一定程度上可以提高g-C3N4的光催化活性.
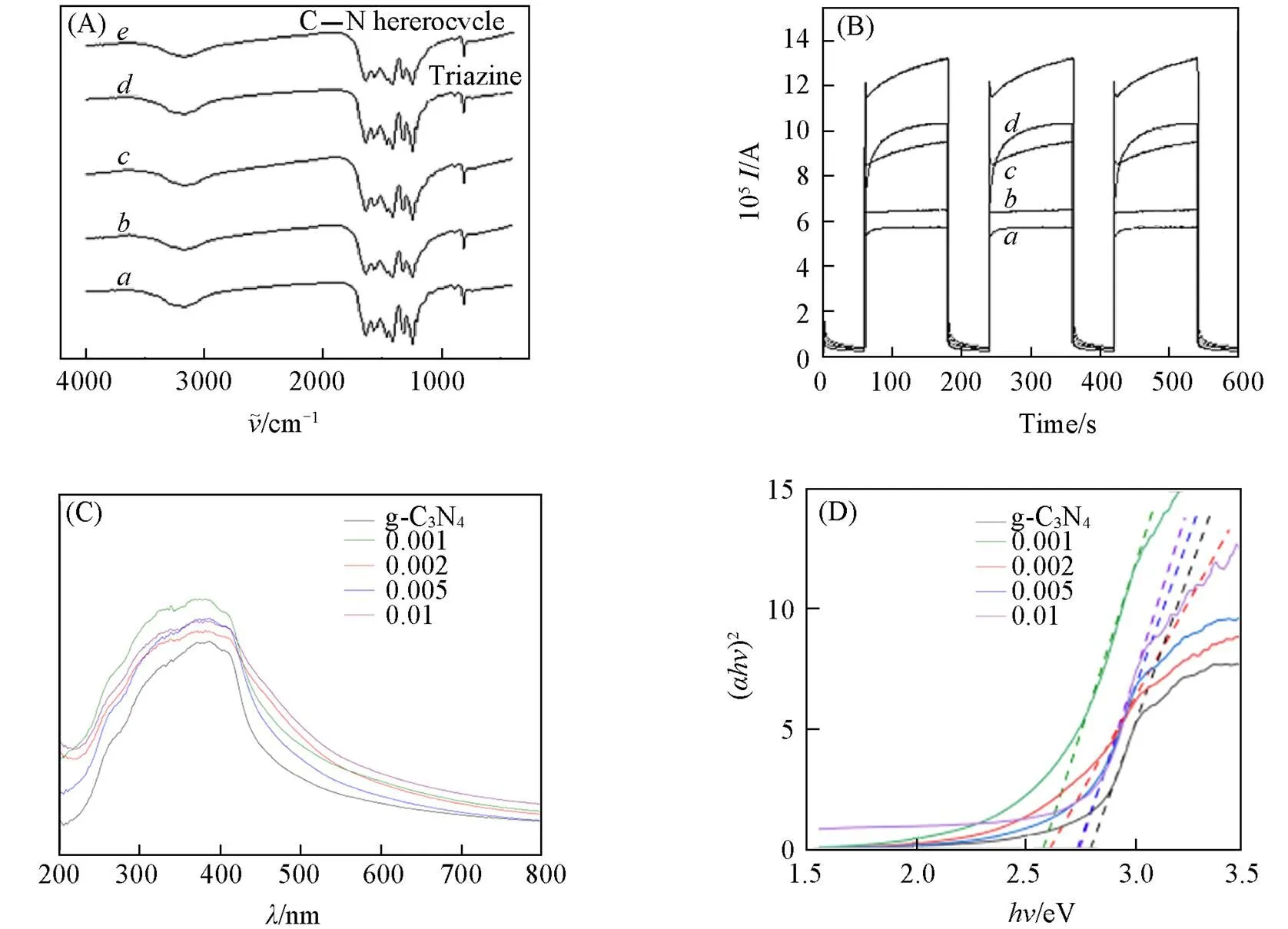
Fig.5 FTIR spectra(A) of g⁃C3N4 powder doped with the different concentrations of K+, photocurrent response curves(B), UV⁃Vis spectra(C) and (αhv)2vs. hv plots(D) of g⁃C3N4 thin film electrode doped with the different concentrations of K+
(A, B)g⁃C3N4; K+concentration/(mol·L-1):. 0.01;. 0.005;. 0.002;. 0.001.
2.2 K+/g-C3N4薄膜电极光电催化DCF的影响因素
图6(A)显示了不同反应条件下DCF的去除率. 可见, 黑暗条件下, DCF的浓度几乎没有变化, 去除率仅为0.32%, 说明g-C3N4薄膜电极对于DCF的吸附作用几乎没有. K+/g-C3N4薄膜电极单独光催化DCF的去除率为14.12%. 当K+/g-C3N4被光照时, 可产生光生电子和空穴, 光生电子可以产生自由基来间接降解DCF, 而光生空穴可以传输到表面直接氧化DCF, 因而K+/g-C3N4薄膜具有一定的光催化活性[17]. K+/g-C3N4薄膜的光催化活性较低, 主要还是由于光生电子和光生空穴的复合率较高. K+/g-C3N4薄膜电极单独电化学氧化DCF的去除率为10.83%, 这表明ITO上的K+/g-C3N4薄膜也有电催化活性, 但催化效率很低. 文献[13]中也报道了ITO上g-C3N4薄膜具有电催化活性. K+/g-C3N4薄膜电极的电催化活性有限, 主要是由于K+/g-C3N4的电导率偏低.

Fig.6 Removal rates of DCF in aqueous solution in the different reaction system(A), effect of voltage(B) and initial pH value(C) on the photoelectrocatalytic degradation of DCF

式中:(mg/L)为反应目标污染物的浓度;(min-1)为反应速率常数;0(mg/L)为初始浓度;(min)为反应时间.
根据一级反应动力学, DCF的降解速率与浓度和反应时间有关, 对公式求导, 得到一级动力学方程为-=ln(1/0). 实验中, DCF的初始浓度为10 mg/L, 根据不同降解时间DCF的浓度, 得到一级动力学参数(表1). 根据表1, K+/g-C3N4薄膜电极光催化(PC)氧化和电化学(EC)氧化的反应速率常数分别为0.0012和0.0008 min-1, 而光电催化(PEC)氧化的反应速率常数为0.0041 min-1, 远远大于两者相加, 可以说明光电催化氧化可以提高对DCF的降解速率.
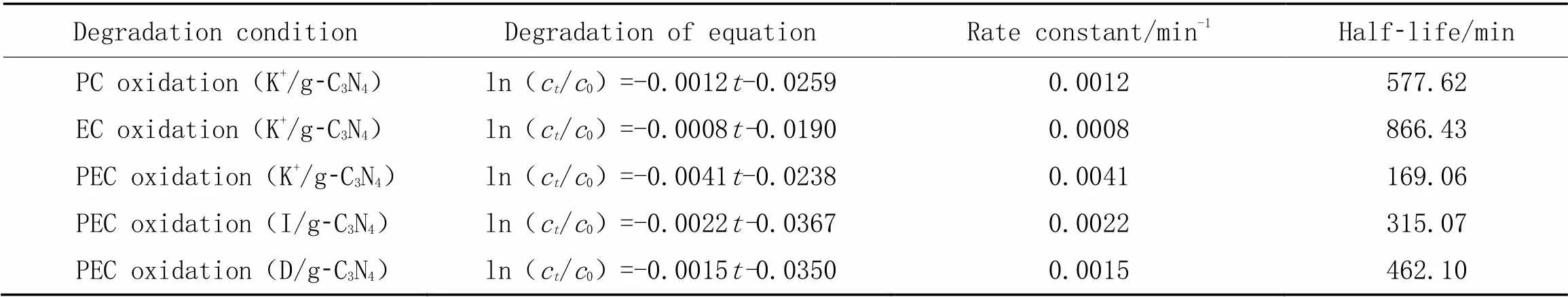
Table 1 First order kinetics parameters of DCF degradation in different conditions*
*PC: Photocatalysis; EC: electrochemistry; PEC: photoelectrocatalysis.
K+/g-C3N4薄膜电极光电催化氧化反应2 h后, DCF的去除率达到39.73%, 是纯I/g-C3N4薄膜电极的1.62倍[图6(A)], 一方面是因为K+的掺杂增加了g-C3N4的负载量, 另一方面是K+的掺杂提升了电极的光电响应,并提高了g-C3N4光生电子和空穴的分离效率以及迁移速率. 从图6(A)还可以看出, K+/g-C3N4薄膜电极和I/g-C3N4薄膜电极光电催化性能明显高于D/g-C3N4薄膜电极, 这主要是由于这两个电极在保证良好的透光性能的同时, 拥有更高的g-C3N4负载量, 因而有更多的催化活性位点, 更有利于光电催化反应的发生.
图6(B)显示当施加电压由0.5 V逐渐增加到2.0 V时, DCF的去除率先升高后降低, 施加电压在1.0 V时, DCF去除率高达40%. 光电催化氧化DCF的反应符合准一级动力学(表2), 电压为1.0 V时, 反应速率常数最大, 达到0.0035 min-1; 当电压大于1 V时, 电压增大反应速率下降, 1.5 V时的反应速率常数与0.5 V时相同, 为0.0020 min-1; 2.0 V时反应速率常数减小至0.0015 min-1. 光电催化氧化过程中, 电压使K+/g-C3N4的能带弯曲, 改变了空间电荷层的厚度, 是影响降解效果的重要因素[19,20]. 由于施加的电压会使K+/g-C3N4的能带弯曲, 增大空间电荷层的厚度, 光生电子-空穴的复合时间延长, 分离的效率提高, 促进了对DCF的降解. 当电压增大到1.0 V时, 空间电荷层的厚度接近薄膜电极的厚度, 光生电子-空穴对充分分离, 形成饱和光电流, DCF的降解效率达到最大. 但是, 当电压继续升高, 超过1.0 V时, 较大的电压会破坏K+/g-C3N4在ITO上的附着, 造成K+/g-C3N4薄膜脱落, 局部的空间电荷层变窄, 光生载流子复合率增大, 导致DCF的去除率降低. 在高电压条件下g-C3N4薄膜会脱落, 是 K+/g-C3N4薄膜电极的主要缺陷. 因此, 采用K+/g-C3N4薄膜电极进行光电催化氧化降解水中有机物污染物时, 施加的电压应该维持在一个相对较低的水平.

Table 2 First order kinetics parameters of DCF degradation in different bias
图6(C)显示, 随着溶液初始pH的下降, K+/g-C3N4薄膜电极光电催化氧化DCF的去除率逐渐升高, 当溶液初始pH为4时, 降解率达到了最大(70%), 降解DCF的速率常数增加到0.0088 min-1(表3). 说明当溶液初始为酸性条件时, 有利于DCF的降解. 从DCF的结构可以看出, 溶液在低pH时, 双氯芬酸中的亚氨基键将会产生质子化形式的—NH3+; 而且, 双氯芬酸中羧基基团在低pH时以羧酸根的形式存在, 而在高pH 时, 却以—COO—的形式存在. 双氯芬酸的酸度系数(pa)大约在(4.35±0.2)左右, 而当pH值在pa值附近时, 双氯芬酸主要以两性离子形式存在, 而两性离子的光解速率最大[21]. 当pH值升高时, 降解率降低, 这是由于, 与两个苯环链接的—NH随着pH值升高, 其质子化程度降低. 而质子化越低, C—N—C越不容易断裂, 而C—N—C的断裂意味着双氯芬酸的降解[22,23]. 因此, K+/g-C3N4薄膜电极光电催化氧化DCF的最佳初始pH为4.
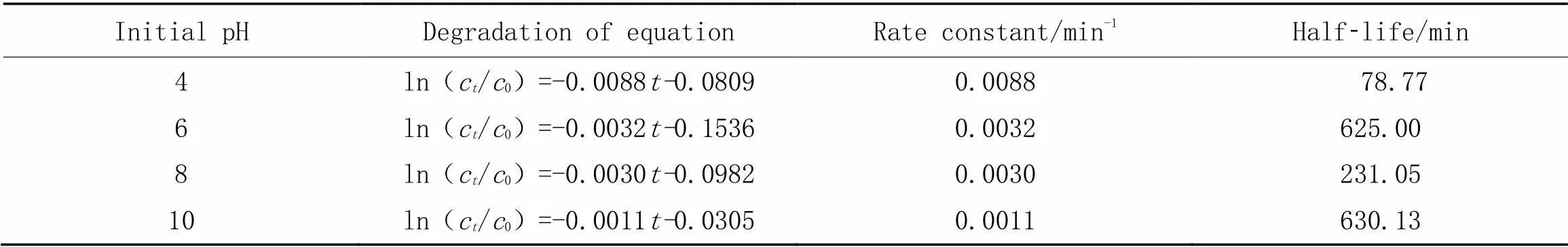
Table 3 First order kinetics parameters of DCF degradation in different conditions of pH
2.3 K+/g-C3N4薄膜电极光电催化氧化DCF的机理
K+/g-C3N4薄膜电极光电催化氧化DCF的速率, 分别是单独光催化和电化学氧化的3.42倍和5.13倍. 在K+/g-C3N4薄膜电极光电催化氧化体系中, 光催化氧化与电化学氧化之间存在相互增强作用, 反应机理如Scheme 1所示. 一方面, 施加的电压可以减少光生电子和光生空穴的复合, 促进光生电荷载流子的分离, 并最终提高光催化氧化降解DCF的速率. 另一方面, 可见光照射产生的光生电子和光生空穴可以消除低电压条件下电极表面的钝化, 也可以产生活性物质提升高电压下电化学氧化降解DCF的速率.
为了阐明活性物质在光电催化降解DCF实验中的作用, 在光电催化降解过程中分别加入了h+, ·OH和·O2-的猝灭剂草酸铵(AO)、 叔丁醇(TBA)和对苯醌(-BQ). 由图7可知, 加入-BQ的时候, 降解动力学几乎不被影响, 说明·O2-对DCF的降解没有作用. 然而, 加入AO和TBA的时候, DCF的降解速率从4.10×10-3min-1分别降到了1.56×10-3和1.12×10-3min-1, 说明在DCF的降解实验中, h+和·OH起主要作用. g-C3N4被可见光辐射产生光生空穴和光生电子, h+可以直接氧化DCF, 也可以和H2O反应生成·OH; ·OH具有强氧化性, 可以攻击DCF环上与—NH—键处于对位的具有亲电子属性的C原子. 另一方面, 施加的电压可以减少光生电子和光生空穴的复合, 促进光生电荷载流子的分离, 增加体系中h+和·OH的数量, 提高降解DCF的速率. 文献[13]也报道了g-C3N4薄膜电极光电催化氧化降解水中苯酚过程中, 光催化氧化和电化学氧化之间存在着协同降解作用. 因此, K+/g-C3N4薄膜电极光电催化氧化过程中, 光催化氧化和电化学氧化之间能够相互增强.
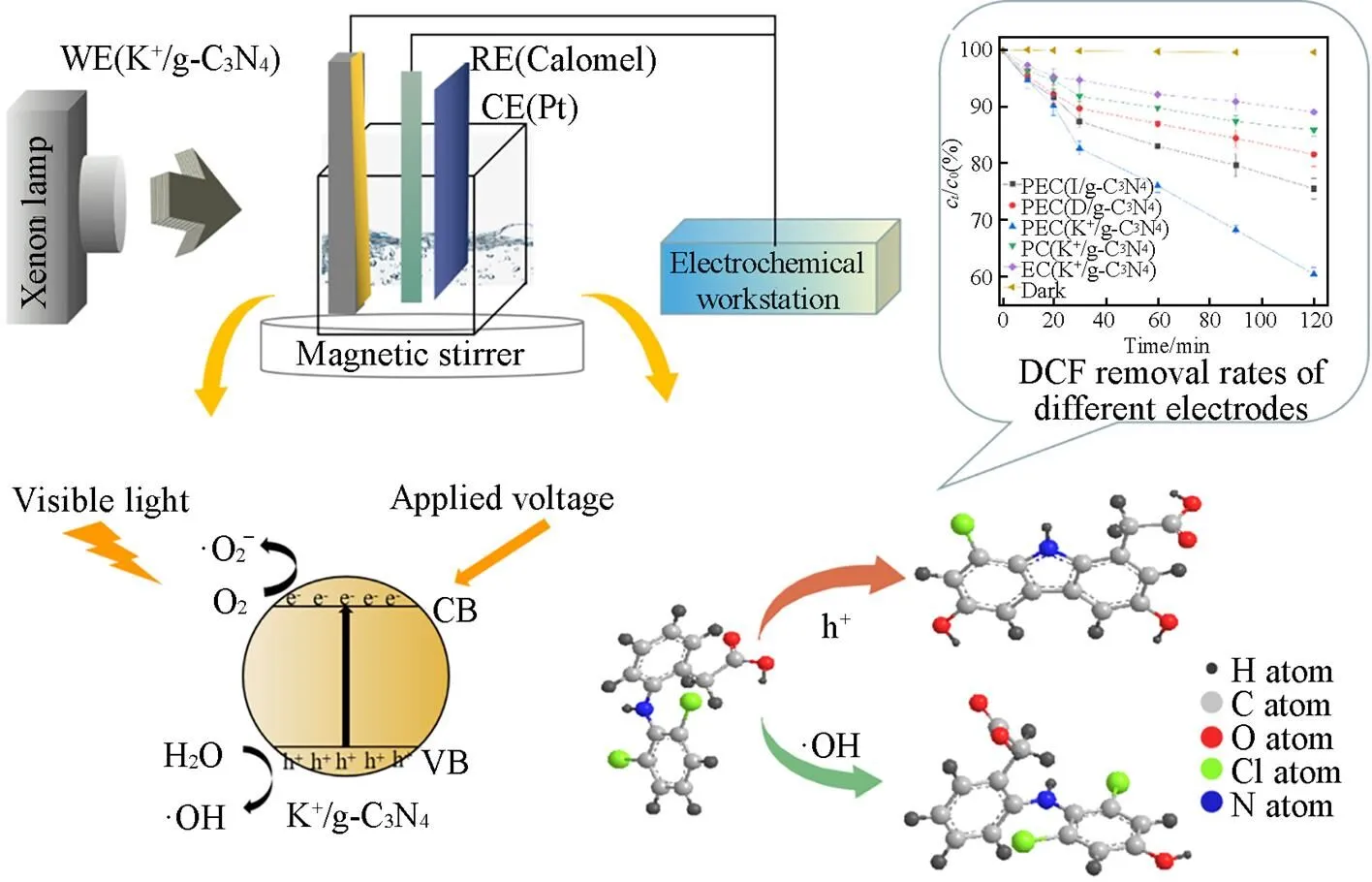
Scheme 1 Mechanism on the PEC degradation of DCF using K+/g⁃C3N4film electrode
WE: working electrode; RE: reference electrode; CE: counter electrode.

Fig.7 Effects of different quenchers in PEC reaction
AO: 1 mmol/L;TBA: 0.5 mmol/L;cBQ: 0.1 mmol/L.
2.4 K+/g-C3N4薄膜电极光电催化氧化DCF的中间产物
针对K+/g-C3N4薄膜电极光催化氧化或电化学氧化降解水中DCF的降解产物, 采用HPLC-MS的方法进行了检测分析, 但是没有检测到中间产物, 可能是由于反应过程中生成的中间产物浓度非常低, 这也说明了单纯的光催化氧化或电化学氧化对水中DCF的降解效率偏低.
针对K+/g-C3N4薄膜电极光电催化氧化降解水中DCF的降解产物, 采用HPLC-MS的方法进行了检测分析, 检测出的中间产物的/值有325, 311, 296, 291, 261, 168, 161, 128和126, 基于检测出的中间产物, 推断出降解路径(Scheme 2). 在光电催化氧化降解过程中, 由于光催化氧化和电化学氧化能够相互加强, 产生的h+和·OH浓度更高. 在这两种活性物质作用下, DCF溶于水后电离产生双氯酚酸阴离子可发生两种反应: (1) 被h+氧化生成咔唑衍生物; (2) 与·OH发生加成反应, 生成多羟基芳香化合物, 最后开环生成小分子物质.
降解反应初始, h+是主要的活性物质, 连接在—NH—邻位的—Cl断裂, 发生脱氯反应, 并在两个苯环之间生成一个稳定的五环结构, 生成咔唑及其衍生物(如产物=291), 这类中间产物稳定性很高, 不易被氧化降[24~26].
DCF环上与—NH—键处于对位的C原子具有亲电子属性, 易被·OH攻击, 生成中间产物/=311. 继续被·OH氧化, 生成含醌的化合物, 同时电子重新排布, —NH—生成—N=C键, 生成中间产物=325[27~29]. —NH—键中的N原子电负性高, 在·OH攻击下断裂, 生成单苯环化合物, 这个反应很迅速, 生成中间产物/=261. 这条路径与文献[24]中描述一致.
同时, —NH—键中的N原子电负性较高, 易被强氧化性的活性物质攻击, 在反应过程中发生断裂, 生成中间产物=161和=168. —Cl和—CH2COOH是吸电子基团, 在光电催化降解的过程中也会被攻击[30]. 中间产物/=161的一个—Cl从苯环上断裂, 被—OH取代, 生成产物/=128; 而中间产物/=168的—CH2COOH被—OH取代, 生成产物/=126.

Scheme 2Degradation pathway of DCF in the photoelectrocatalytic reaction
从图8(A)~(E)可以看出, K+/g-C3N4薄膜电极在重复使用5次后, 对降解DCF仍表现出较高的活性. 这说明K+/g-C3N4与ITO玻璃之间的黏附性较强, K+/g-C3N4薄膜电极在整个光电体系中具有良好的稳定性. 在过去的几十年里, 低效率限制了光催化技术的实际应用, 而高能耗限制了电化学技术的实际应用. 由于K+/g-C3N4薄膜电极具有优异的光电催化活性和稳定性, 在光催化和电化学反应的协同作用下, 这一情况有望得到改善.

Fig.8 Stability test of K+/g⁃C3N4 film electrode for photoelectrocatalytic degradation of DCF
(A) First; (B) second; (C) third; (D) fourth; (E) fifth.
3 结 论
本文通过原位煅烧法和掺杂K+制备了兼具高光学质量和良好电化学性能的K+/g-C3N4薄膜光电极, 利用该电极光电催化氧化降解水中DCF的最佳反应参数为: K+掺杂浓度为0.002 mol/L, 双氯芬酸钠初始pH为4, 电压为1 V, 光源强度为0.96 W/cm2, 反应2 h, 去除率达到70%. K+/g-C3N4薄膜电极光电催化氧化过程中, 光催化氧化和电化学氧化之间存在协同作用, 两者相互增强, 并提高了反应过程中h+和·OH浓度, 强化了水中DCF光电催化氧化降解, DCF既可能被h+氧化生成咔唑衍生物, 也可能被 ·OH氧化成小分子有机物. 本研究能够促进光催化剂g-C3N4在环境水处理中的应用.
[1] Chen C. C., Ma W. H., Zhao J. C.,.,2010,(11), 4206—4219
[2] Xiao X., Zhang W. D.,,2011,(3), 287—291
[3] He Y. L., Zhang X., Wei Y. Z., Chen X. Y., Wang Z. M., Yu R. B.,.,2020,(3), 447—452
[4] Wang H. L., Pang L., Jiang W. F., Yang C.,.,2012,(9), 1071—1073
[5] Shao W. C., Jia G. Y.,.,2014,(12), 2101—2107
[6] Li Q., Zhang N., Yang Y., Wang G. Z., Ng D. H. L.,,2014,(29), 8965-8972
[7] Liang R. Y., Xu D. D., Zha W. Y., Qi J. Z., Huang L. H.,, 2016,(11), 1953—1959(梁瑞钰, 徐冬冬, 查文莹, 齐楫真, 黄浪欢. 高等学校化学学报2016,(11), 1953—1959)
[8] Phang S. J., Goh J. M., Tan L. L., Lee W. P. C., Ong W. J., Chai S. P.,.,2021,(4), 4388—4403
[9] Li L., Qi Y., Hu J., An W., Lin S., Liang Y., Cui W.,.,2015,, 278—281
[10] Liu C., Dong X. L., Hao Y. C., Wang X. Y., Ma H. C., Zhang X. F.,.,2017,(20), 11872—11880
[11] Tong Z. W., Yang D., Shi J. F., Nan Y. H., Sun Y. Y., Jiang Z. Y.,., 2015,(46), 25693—25701
[12] Patra P. C., Mohapatra Y. N.,.,2021,, 618—622
[13] Liang F. F., Zhu Y. F.,.,2016,, 324—329
[14] Xu J., Brenner T., Chabanne L., Neher D., Antonietti M., Shalom M.,.,2014,(39), 13486—13489
[15] Cao D. D., Lv R., Yu A. C.,2019,(4), 442—450(曹丹丹, 吕荣, 于安池.2019,(4), 442—450)
[16] Tauc J.,., 1968,(1), 37—46
[17] Yan S. C., Li Z. S., Zou Z. G.,,2010,(6), 3894—3901
[18] Cui Y., Ma Q., Deng X., Meng Q., Cheng X., Xie M., Li X., Cheng Q., Liu H.,, 2017, 206
[19] Wei Z., Liang F., Liu Y., Luo W., Wang J., Yao W., Zhu Y.,.,2017,, 600—606
[20] Ji M., Jin L. N., Fu J. F., Li X. Z.,,2006,(2), 145—148(季民, 金洛楠, 傅剑锋, 李湘中. 环境化学,2006,(2), 145—148)
[21] Liu A. R., Zhao Y. W., Wei Z. Z., Zhang Y. J., Song Q.,.,2017,(4), 4006—4014
[22] Zhang T., Oyama T., Aoshima A., Hidaka H., Serpone N.,,2001,(2), 163—172
[23] Yu Z. Q., Chuang S. S. C.,.,2008,(3/4), 277—285
[24] Espino⁃Estévez M. R., Fernández⁃Rodríguez C., González⁃Díaz O. M., Arana J., Espinós J. P., Ortega⁃Méndez J. A., Dona⁃Rodríguez J. M.,.,2016,, 82—95
[25] Zhang W., Zhou L., Shi J., Deng H.,., 2017,, 167—176
[26] Pérez⁃Estrada L. A., Malato S., Gernjak W., Agüera A., Thurman E. M., Ferrer I., Fernández⁃Alba A. R.,.,2005,(21), 8300—8306
[27] Maier M. P., Corte S. D., Nitsche S., Spaett T., Boon N., Eisner M.,l., 2014,(4), 2312—2320
[28] Forrez I., Carballa M., Verbeken K., Vanhaecke L., Ternes T., Boon N., Verstraete W.,.,2010,(9), 3449—3954
[29] Xu Z., Hou Y., Liu H., Qiang Z., Qu J.,,2009,(17), 4172—4179
[30] Liu Q., Zheng Z., Luo X. Z., Zhang J. B., Zheng B. G.,.,2011,(10), 1700—1704(刘群, 郑正, 罗兴章, 张继彪, 郑宾国. 环境化学, 2011,(10), 1700—1704)
Preparation of Potassium Doped g-C3N4Thin Film Photoanode and Its Application in Photoelectrocatalytic Oxidation of Diclofenac Sodium in Water
GONGYanxi, WANGJianbing*, CHAIBuyu, HANYuanchun, MAYunfei, JIAChaomin
(,,100083,)
The difficulty in preparing thin film photoelectrodes with high optical quality and excellent electro- chemical property limited the application of photoelectrocatalytic oxidation process in water treatment. In this paper, g-C3N4thin film electrodes loaded on indium tin oxide(ITO) glass was prepared bycalcination method and used potassium doping to improve their photoelectrocatalytic activity. The electrodes were characterized, the photoelectrocatalytic degradation of diclofenac sodium(DCF) in water with them was studied, and the DCF degradation pathways were investigated. The results showed that thecalcination method could prepare high-quality K+/g-C3N4thin film photoelectrodes, and the doping of K+insignificantly changed the crystalline shape, valence state, and porous morphology of g-C3N4on the electrode. However, it could increase the loading of g-C3N4on ITO glass and enhance the photocurrent response of the electrode to visible light. The optimal doping concentration of K+was 0.002 mol/L. The rate constant of DCF degradation in photoelectrocatalytic oxidation process with the K+/g-C3N4thin film electrode was 1.86 times higher than that with the pure g-C3N4film electrode. The DCF removal rate from water in 2 h reaction time reached 70% with the initial pH of 4, applied potential of 1 V, and light intensity of 0.96 W/cm2. For the photoelectrocatalytic oxidation of DCF with the K+/g-C3N4film electrode, there was a synergy between photocatalytic oxidation and electrochemical oxidation. They could optimize each other and enhance the concentration of the photogenerated holes(h+) and hydroxyl radicals(·OH) produced in the reaction process. Under the action of these two active substances, DCF in water was oxidized by h+into carbazole derivatives, reacted with ·OH to form polyhydroxy aromatic compounds, and finally occurred reaction of ring opening to form small molecules.
Potassium doped g-C3N4thin film photoanode; Photoelectrocatalytic oxidation; Diclofenac sodium; Photocatalytic oxidation; Electrochemical oxidation
O643
A
10.7503/cjcu20220005
2022-01-04.
2022-03-10.
王建兵, 男, 博士, 教授, 主要从事水环境高级氧化技术方面的研究. E⁃mail: wangjb@cumtb.edu.cn
国家自然科学基金(批准号: 51978658)资助.
the National Natural Science Foundations of China(No.51978658).
(Ed.: V, K, S)
猜你喜欢
——潘桂棠光生的地质情怀
沉积与特提斯地质(2021年2期)2021-07-20 06:33:26
陶瓷学报(2019年5期)2019-01-12 09:17:34
科技创新导报(2017年18期)2017-09-09 07:19:21
三峡大学学报(自然科学版)(2017年1期)2017-03-20 15:30:23
物理化学学报(2017年3期)2017-03-11 00:25:30
化工管理(2016年21期)2016-03-14 06:36:51
昭通学院学报(2016年5期)2016-02-24 10:51:12
中国资源综合利用(2016年9期)2016-01-22 08:35:22
物理化学学报(2015年5期)2015-02-28 17:35:02
石家庄铁道大学学报(自然科学版)(2015年3期)2015-02-28 15:05:43
