
动物源性食品中β-受体激动剂残留检测的方法验证
2022-07-18 03:51孙秀菊郭艳琼王建军马占山何建东
现代农业科技 2022年13期
郭 琳 孙秀菊 郭艳琼 梁 海 王建军 马占山 何建东
(1包头市农牧科学技术研究所,内蒙古包头 014000;2包头市农畜产品质量安全中心,内蒙古包头 014000)
方法验证是指实验室通过核查,提供客观、有效的证据证明满足检测方法规定的要求[1]。方法验证是保证结果准确性和可靠性的前提和基础,实验室在引用和使用标准方法之前,应认真研究标准方法中的内容和要求,如人员、设备设施、场所环境等条件。此外,还应通过试验验证实验室是否有执行该标准方法的技术能力[2-6]。
β-受体激动剂又称“瘦肉精”,人如果食用含有β-受体激动剂残留的动物组织,当残留量达到一定程度时就会对人体造成毒副作用,甚至急性中毒。自1997年开始,我国就将近有20种β-受体激动剂列入养殖禁用名单。因此,为保障食品安全,对动物源性食品中β-受体激动剂残留的准确检测具有重大意义。本文以《动物源性食品中β-受体激动剂残留检测 液相色谱-串联质谱法》(农业部1025号公告-18-2008)为基础,对特布他林、西马特罗、沙丁胺醇、氯丙那林、莱克多巴胺、喷布特罗、克仑特罗等7种化合物进行方法优化验证,验证主要内容包括线性范围、方法检出限、方法定量限、正确度、精密度。
1 材料与方法
1.1 主要仪器和试剂
主要仪器有液相色谱-串联质谱仪(Agilent1260-6420)、恒温水浴振荡器、高速离心机、酸度计、涡旋振荡器、全自动氮吹仪。
主要试剂有特布他林、西马特罗、沙丁胺醇、氯丙那林、莱克多巴胺、喷布特罗、克仑特罗标准品;乙酸乙酯、叔丁基甲醚、甲醇,均为色谱级;β-盐酸葡萄糖醛苷酶/芳基硫酸酯酶,2%甲酸水溶液,3%氨水甲醇溶液,0.2 mol/L乙酸铵缓冲液,0.1 mol/L高氯酸溶液,10 mol/L氢氧化钠溶液。
1.2 试验方法
1.2.1 前处理过程。称取2.00 g肉样于50 mL离心管中,加入8.0 mL 0.2 mol/L乙酸铵缓冲液(pH值=5.2)、40 μL β-盐酸葡萄糖醛苷酶和芳基硫酸酯酶,涡旋混匀,于37℃下避光水浴振荡16 h。取出冷却至室温之后,涡旋混匀,在10 000 r/min条件下高速离心10 min,然后将上清液转移至另一个50 mL离心管中,加入5 mL 0.1 mol/L高氯酸溶液,涡旋混匀,用高氯酸调节pH值至1.0±0.2,在10 000 r/min条件下高速离心10 min,将上清液转移至另一个50 mL离心管中,用10 mol/L氢氧化钠溶液调节pH值至9.5±0.2,加15 mL乙酸乙酯,涡旋混匀,振荡10 min,在5 000 r/min条件下离心5 min,移取上层有机相至另一个50 mL离心管内。在下层水相中加入10 mL叔丁基甲醚,涡旋混匀,并振荡10 min,在5 000 r/min条件下离心5 min,合并有机相,50℃下氮气吹干,用5 mL 2%甲酸溶液溶解,备用。
取全部备用液过经甲醇、水、2%甲酸溶液各3 mL活化的MCX固相萃取柱,再依次使用2%甲酸溶液、甲醇各3 mL淋洗,抽干,用2.5 mL 3%氨水甲醇洗脱,将洗脱液在50℃下氮气吹干。残余物用0.2 mL甲醇-0.1%甲酸溶液(10 mL+90 mL混合)溶解,涡旋混匀,15 000 r/min高速离心10 min后,经0.22 μm滤膜过滤后上机待测[7]。
1.2.2 仪器条件。色谱条件:色谱柱为Eclipse Plus C18(100 mm×2.1 mm×3.5μm);流动相 A 为 0.1%甲酸水,流动相B为0.1%甲酸甲醇;柱温为30℃;进样量为10 μL;流速 0.3 mL/min。 梯度洗脱程序:0 min,97.0%流动相 A,3.0%流动相 B;0.50 min,97.0%流动相 A,3.0%流动相 B;2.00 min,50.0%流动相 A,50.0%流动相 B;2.10 min,10.0%流动相 A,90.0%流动相 B;7.50 min,10.0%流动相 A,90.0%流动相 B;7.60 min,97.0%流动相 A,3.0%流动相 B;16.00 min,97.0%流动相A,3.0%流动相B。质谱:电喷雾离子源正离子扫描;多反应监测。干燥气温度300℃,流速13 L/min。
1.2.3 方法验证过程。具体包括以下几方面的内容。
(1)线性范围。称取2.00 g空白试样,分别添加7种β-受体激动剂混标工作液,制得浓度为0、0.25、0.50、1.00、2.00、5.00 μg/kg 的系列标准工作液,按照前述前处理过程进行处理,过0.22 μm滤膜备用。
(2)方法检出限、定量限。以信噪比等于3的标液浓度作为检出限,以信噪比等于10的标液浓度作为定量限,该标准方法中规定7种化合物检出限为0.25 μg/kg、定量限为 0.5 μg/kg。 称取 2.00 g 空白样品,添加检出限和定量限浓度水平目标化合物,按照上述前处理步骤处理后,备用,待上机检测。
(3)正确度。采用加标回收方式验证正确度。β-受体激动剂类是禁用药,针对食品中的禁用物质,回收率应在方法测定低限、两倍方法测定低限和十倍方法测定低限进行3个水平试验[2]。根据方法中给出的检出限和定量限,依次添加 0.25、0.50、2.50 μg/kg 3个水平浓度,进行加标回收试验。
(4)精密度。准确称取2.00 g空白试样,分别添加7种β-受体激动剂混标工作液至加标量为标准曲线中间浓度,本试验以添加量为0.5 μg/kg为例,按照前处理过程操作,上机平行测定6次,计算相对标准偏差。
2 结果与分析
2.1 色谱条件优化
在Eclipse Plus C18色谱柱上,对比了0.1%甲酸乙腈和0.1%甲酸甲醇作为流动相对分离效果的影响。结果表明,在Eclipse Plus C18色谱柱上,0.1%甲酸甲醇的分离度优于0.1%甲酸乙腈,0.1%甲酸水溶液的分离度优于纯水。因此,选用0.1%甲酸水溶液-0.1%甲酸甲醇作为流动相进行梯度洗脱。
2.2 质谱条件优化
母离子优化,即分别对每一种单标溶液进行全质量数扫描,得到化合物的准分子离子。子离子优化,即对其母离子进行电子轰击,得到碰撞产生的子离子,选择2个响应值比较高的子离子,与母离子组成2组特定离子对,作为该化合物的定量离子对和定性离子对。毛细管电压和碰撞能量优化,即分别对7种目标化合物施加毛细管电压70~160 V,以5 V为梯度进行优化,在最佳毛细管电压条件下,对每一对离子对设置碰撞能量5~50 eV,以3 eV为梯度进行优化,分别得到7种目标化合物的最佳毛细管电压和碰撞能量。目标化合物的定性离子、定量离子以及碰撞电压和能量见表1。
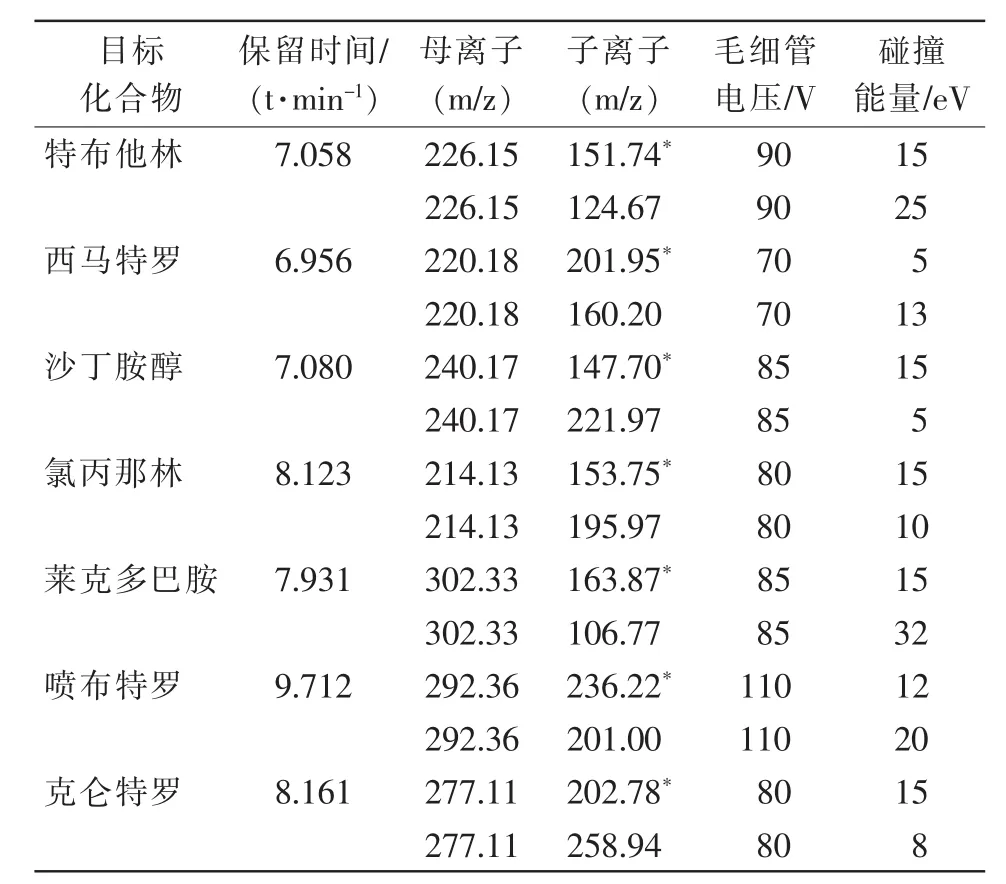
表1 7种β-受体激动剂的毛细管电压和碰撞能量
2.3 工作曲线及线性范围
将处理好的系列标准溶液按照浓度由低到高的顺序供液相色谱-串联质谱测定,以各标准溶液的浓度为横坐标、各标准物质定量离子色谱峰面积为纵坐标绘制标准曲线,如图1~7所示。7种标准物质溶液的标准曲线相关系数满足《实验室质量控制规范食品理化检测》(GB/T 27404—2008)中相关系数不应低于0.99的要求[3]。
2.4 方法检出限与定量限
由表2可知:7种目标化合物的检出限信噪比(S/N)为 28.2~636.7,均大于 3;定量限信噪比(S/N)为62.7~2 972.1,均大于10。说明在现有条件下该方法的检出限和定量限可以满足标准方法中规定的检出限 0.25 μg/kg 和定量限 0.5 μg/kg。
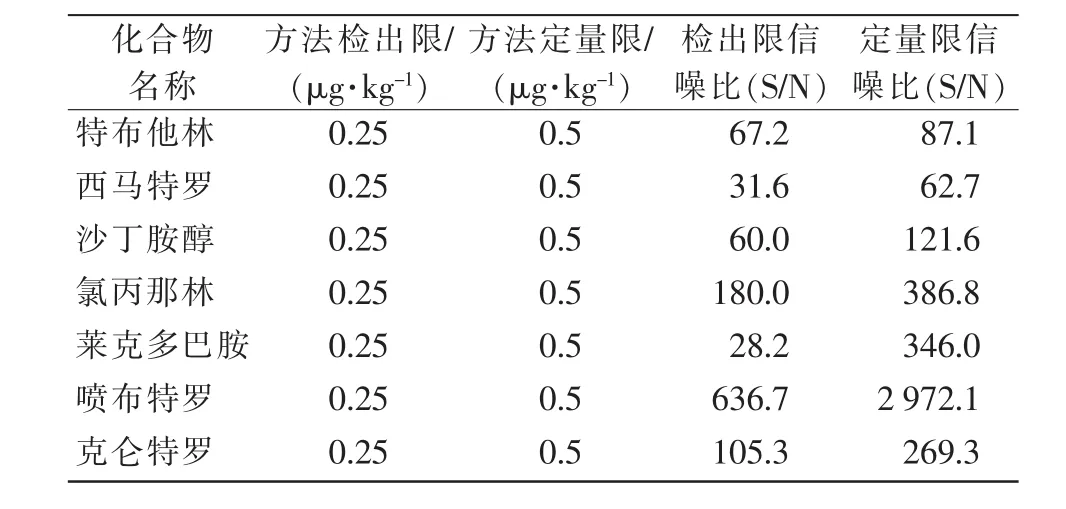
表2 方法检出限、定量限数据
2.5 正确度
以空白样品为本底,添加7种不同浓度β-受体激动剂,其回收率范围为73.2%~102.4%,加标回收试验结果见表3。满足标准方法中的要求,即以空白添加标准校正,其回收率范围为70%~120%。
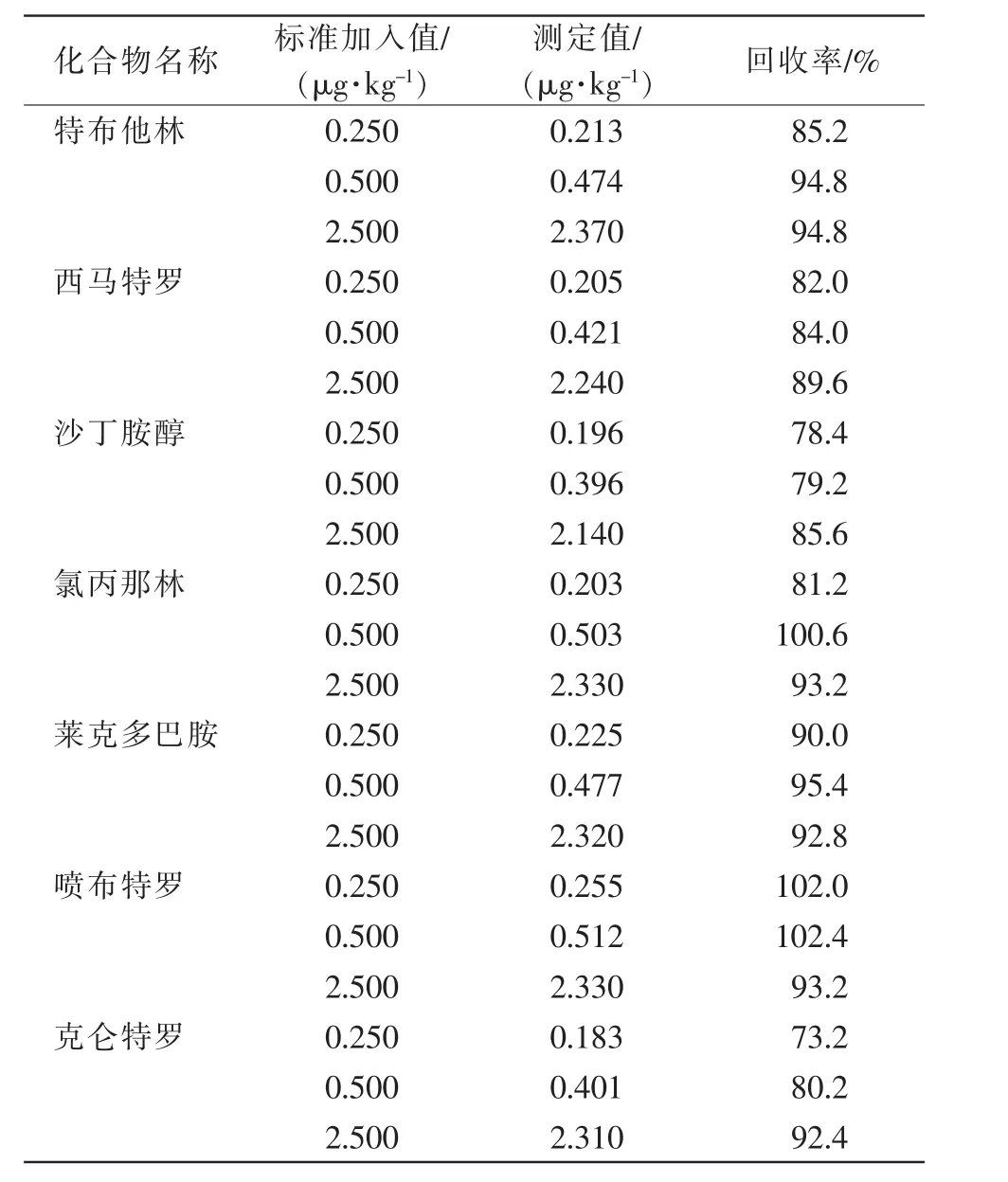
表3 试样加标回收率测定数据
2.6 精密度
由表4可知,在空白样品中添加7种β-受体激动剂进行6次重复试验,计算相对标准偏差结果为6.6%~11.4%,满足标准方法中批内相对标准偏差应≤20%的要求。
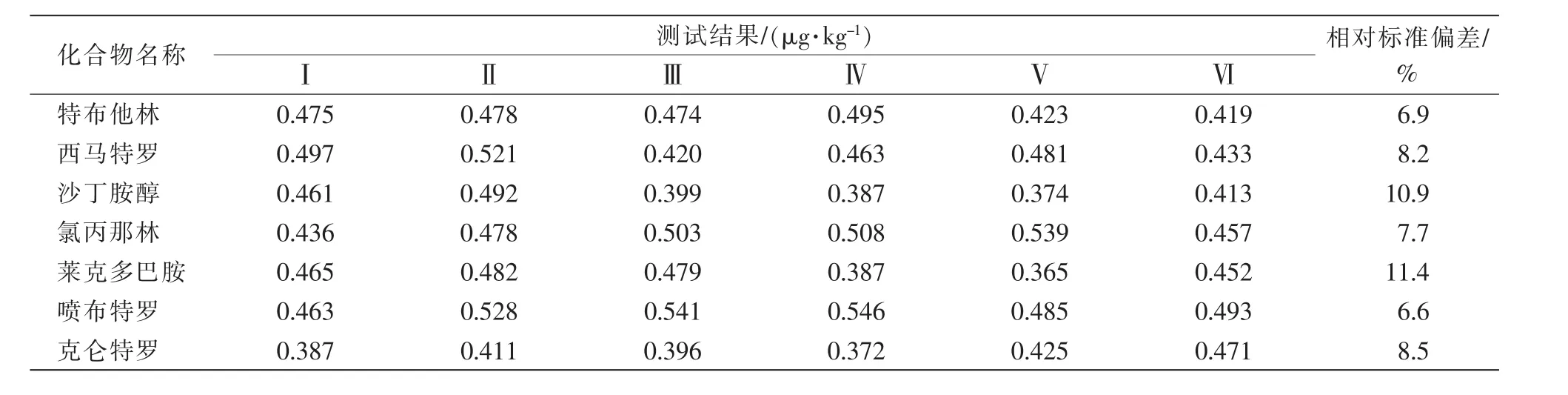
表4 精密度测试数据
3 结论与讨论
本文以《动物源性食品中β-受体激动剂残留检测液相色谱-串联质谱法》(农业部1025号公告-18-2008)为基础,以葡萄糖醛苷酶、芳基硫酸酯酶为酶解剂,用pH值5.2的乙酸-乙酸铵缓冲溶液进行提取,经高氯酸沉淀蛋白后,用氢氧化钠调节pH值至9.5,再分别经乙酸乙酯和叔丁基甲醚萃取后,过MCX固相萃取柱净化,Eclipse Plus C18色谱柱分离,以0.1%甲酸水溶液-0.1%甲酸甲醇为流动相,UPLCMS/MS上机测试,对特布他林、西马特罗、沙丁胺醇、氯丙那林、莱克多巴胺、喷布特罗、克仑特罗等7种化合物进行方法优化验证。结果表明:特布他林、西马特罗、沙丁胺醇、氯丙那林、莱克多巴胺、喷布特罗、克仑特罗等7种目标化合物在0~2.5 μg/kg范围内有良好的线性关系,相关系数达0.99以上,方法检出限、定量限满足方法中的要求,平均回收率73.2%~102.4%,相对标准偏差≤20%。本文建立了畜产品中特布他林、西马特罗、沙丁胺醇、氯丙那林、莱克多巴胺、喷布特罗、克仑特罗等7种β-受体激动剂的液相色谱-串联质谱检测方法的验证过程,证实了在现有的设备设施、人员、环境等条件下有能力正确运用本方法进行检测。
猜你喜欢
——以离子色谱法测定冬瓜中亚硝酸盐含量为例
现代食品(2022年15期)2022-09-08
实用癌症杂志(2022年8期)2022-08-15
波谱学杂志(2022年2期)2022-06-14
安徽农业科学(2022年6期)2022-04-11
四川农业科技(2021年9期)2021-11-19
科学与财富(2021年13期)2021-07-04
生物工程学报(2020年6期)2020-07-31
家庭医药(2020年2期)2020-03-17
科技与创新(2015年24期)2015-12-21
中小企业管理与科技·中旬刊(2014年7期)2014-09-24
