
高尿酸血症患者肠道菌群特征分析
2022-07-14 03:56孙超王健王信谢长好李志军
淮海医药 2022年3期
孙超,王健,王信,谢长好,李志军
高尿酸血症是一种由嘌呤代谢失调引起的常见的代谢性疾病,其特征是血清尿酸水平升高[1],随着人们饮食和生活方式的改变,高尿酸血症的发病率在世界范围内逐年增加。高尿酸血症的患病率在美国已达到21.4%[2],截至2017年底,中国确诊病例已达1.7亿例,中国女性和男性的高尿酸血症发生率分别为7.9%和19.4%[3]。超过30%~40%的尿酸由肠道排泄,肠道清除功能障碍可导致高尿酸血症[4]。肠道是人体最大的内分泌器官之一,体内有数千种微生物群落。肠道菌群和许多代谢疾病有关,比如痛风[5]、糖尿病[6]、肥胖症[7]等。目前,关于HUA病理学和生理学机制的研究比较深入,但肠道菌群与HUA之间作用关系及其机制的研究较少。本研究拟应用16 SrDNA测序技术,来明确HUA患者与正常对照人群之间肠道微生物出现何种改变,为进一步明确肠道菌群失调在HUA的发生发展中的作用机制进行探讨,为HUA的治疗提供思路,为患者合理应用益生菌提供参考。
1 材料与方法
1.1 一般资料 收集2019年9月—2020年9月我院风湿科10例符合纳入标准的HUA患者的粪便样本为HUA组,另外收取HUA组直系亲属10例健康人的粪便为D组。HUA组年龄17~59岁;均符合非同日两次血尿酸的水平超过420 μmol/L的纳入标准[8]。排除标准:(1)有其他自身免疫性疾病及代谢性疾病如系统性红斑狼疮、类风湿性关节炎、系统性硬皮病、痛风、糖尿病、肥胖症等;(2)近1月内使用任何抗生素、益生菌或抗微生物制剂;(3)曾有消化道手术或患有其他消化道器质性疾病。所有患者留样前均为初诊初治,未曾应用激素类及降尿酸药物治疗。健康对照组10名系患者健康直系家属,年龄17~60岁。纳入标准:近1月内未使用任何抗生素、益生菌或抗微生物制剂;消化道未进行过手术;消化道无其他器质性疾病。本研究通过我院医学伦理委员会批准,所有研究对象均签署知情同意书。
1.2 研究方法 采集所有健康人及HUA患者新鲜粪便,使用带勺粪便采集管收集中间段新鲜的粪便标本3~5 g(没有接触空气),尽快将装有样品的采集管置于-80 ℃冰箱保存,保持样品原始状态的完整性。提取样本DNA的方式参照粪便样本DNA提取试剂盒使用方法(QIAamp®DNA Stool Mini®Kit,上海创赛科技有限公司)。采用illumina hiseq平台对细菌16 SrDNA V3~V4区进行高通量测序,其结果进行生物信息学分析。具体如下:(1)进行测序数据的质量控制。测序数据经过质量评估后,过滤掉低质量序列,并直接截取双末端的前240 bp用于后续分析。(2)用DADA2学习序列错误率,去除重复序列,并基于错误模型使用其核心算法进一步做质控。(3)质控后的序列根据重叠部分合并,生成扩增子序列变异体(amplicon sequence variants, ASVs)的丰度表,ASVs进一步做去除嵌合体处理将测序reads聚类得到OTUs,采用高分辨率的DADA2方法来推测核糖体序列变体。(4)根据16S序列的差异,用SILVA v138训练的朴素贝叶斯分类器,给ASVs做物种注释。(5)先用DADA2中的assignTaxonomy函数给每个OTU做注释(门纲目科属水平),再用函数 addSpecies 做种水平注释。另外,用PICRUSt2进一步对ASVs序列做功能注释并得到功能丰度数据。
1.3 统计学方法 用R软件包phyloseq计算α多样性(Alpha diversity)指数(包括Shannon, Simpson, InverseSimpson, Richness和Evenness),用基于Bray-Curtis距离的主坐标分析(Principal coordinates analysis,PCoA)方法来探索样本之间的差异,采用非参检验 Wilcoxon Rank Sum Test法筛选出样本组间差异的菌群及功能,用PERMANOVA方法来检验组间的差异,所有的分析在R统计分析平台上实现。
2 结果
2.1 2组样本间肠道菌群的物种组成 对肠道菌群进行物种分析,共鉴定出8个门,8个属。门水平主要包括Firmicutes,Bacteroidota,Actinobacteriota,Fusobacteriota,Proteobacteria,Verrucomicrobiota,Cyanobacteria等,门水平丰度图见图1。属水平主要包括Bacteroides,Faecalibacterium,Prevotella,Bifidobacterium,Agathobacter,Fusobacterium,CAG-352等,属水平丰度图见图2。这7种主要菌门及主要菌属在这2组之间两两比较差异无统计学意义(P>0.05),各组间的含量有一定的波动,但差异无统计学意义。

图1 2组样本间菌群门水平丰度图

图2 2组样本间菌群属水平丰度图
2.2 2组样本间菌群的多样性分析 2组样本丰富度和多样性主要通过α多样性指数(Alpha diversity)(包括Shannon、Simpson、inverseSimpson、Richness、Evenness)进行组间统计分析,见图3。图3中HUA组的α多样性均较对照组低,但差异无统计学意义(P>0.05)。基于Bray-Curtis距离的β多样性分析显示2组间肠道菌群构成差异无统计学意义(P=0.131,R2=0.067)。见图4。
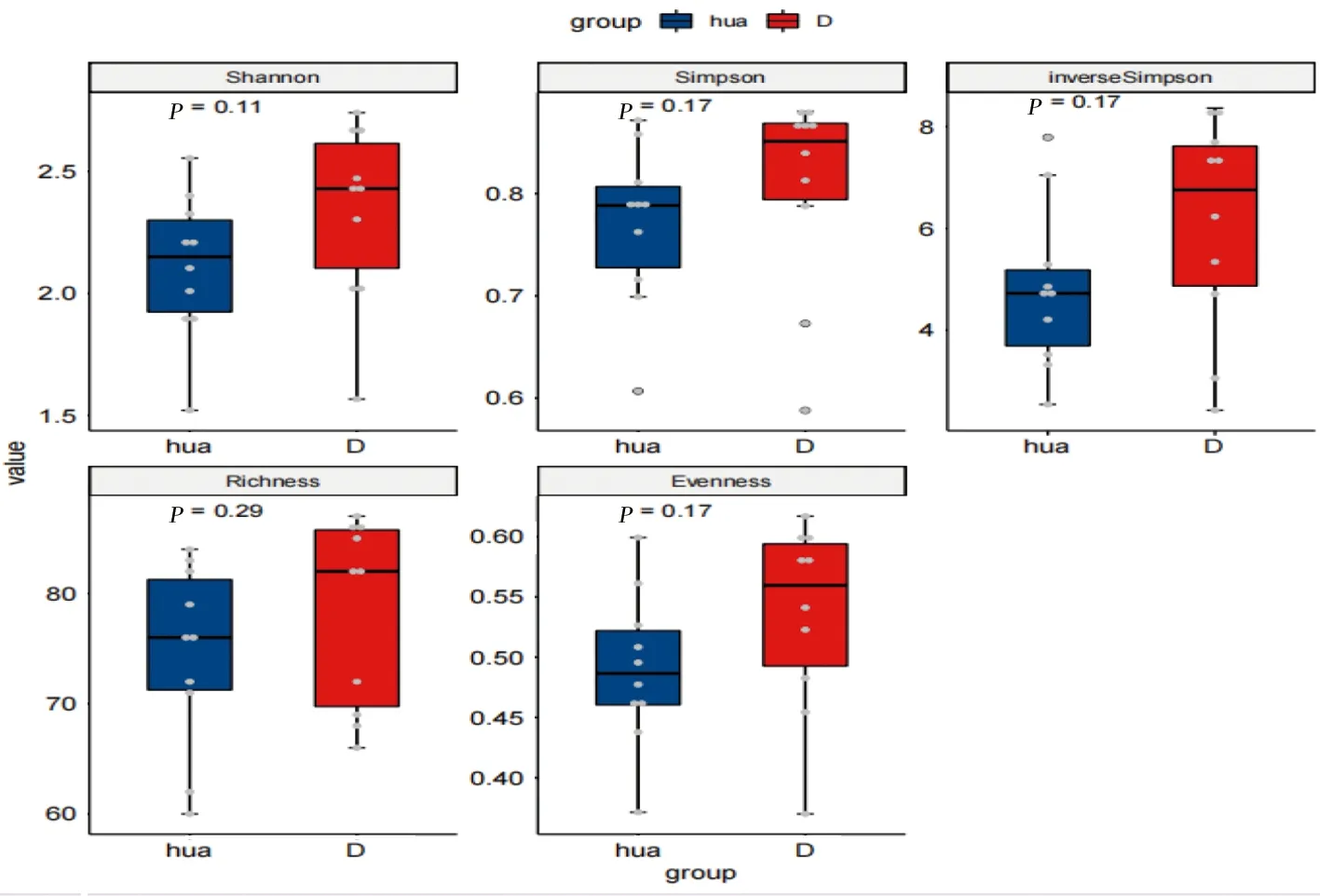
图3 不同alpha指数的组间比较结果
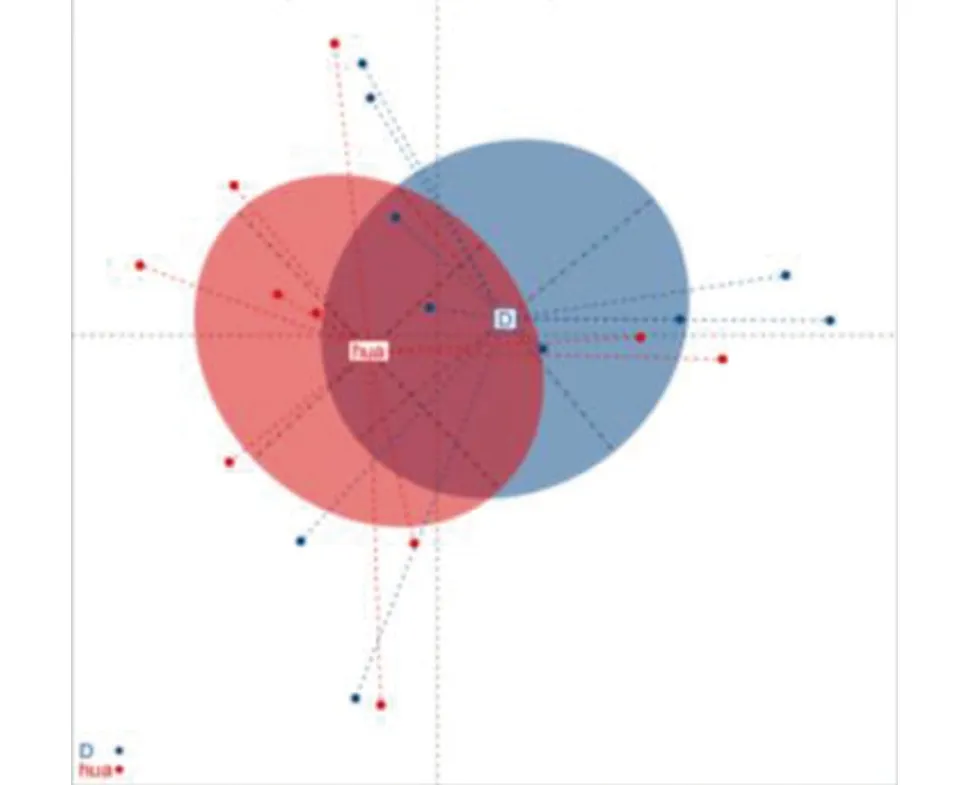
图4 PCoA分析图(β多样性分析结果)
2.3 样本组间差异性分析 用Wilcox方法检验成对样本之间在门、纲、目、科、属水平上菌群相对丰度的差异,属水平上的分析显示Romboutsia、[Clostridium] innocuum group、Christensenellaceae R-7 group、Agathobacter、[Ruminococcus] gnavus group、Clostridium sensu stricto 1、Shuttleworthia、Eisenbergiella在2组间差异有统计学意义(P<0.05)。见表1。

表1 2组样本间细菌在属水平上的比较
3 讨论
HUA是一种复杂的代谢性疾病,与痛风、高血压、糖尿病、肾病等疾病的发生发展密切相关,其对人类的危害是不可忽视的问题。人体胃肠道定植了数万亿个微生物,肠道菌群参与物质和能量的代谢,促进免疫系统的发育和成熟,形成黏膜屏障,保护宿主免受病原体的侵袭。为探索肠道菌群与HUA之间的相关性,本研究对HUA患者和健康对照组人群的肠道菌群进行差异分析。
一些研究[9-10]表明较低的肠道菌群α多样性与多种疾病相关,例如肠易激综合征、肥胖、IBD和干燥综合征等,本研究分析显示HUA患者的物种丰富度低于健康对照组,与既往报告[11]的中国痛风患者的菌群多样性较低一致,表明肠道生态失调,维持更多样化的肠道微生物群组成通常被认为对健康有益。
肠道菌群的差异分析显示Romboutsia、[Clostridium] innocuum group、Christensenellaceae R-7 group、Agathobacter、[Ruminococcus] gnavus group、Clostridium sensu stricto 1、Shuttleworthia、Lachnospira、Eisenbergiella在2组间差异有统计学意义。其中Romboutsia、[Clostridium] innocuum group、[Ruminococcus] gnavus group、Shuttleworthia、Eisenbergiella在HUA组中含量较多,Christensenellaceae R-7 group、Agathobacter、Clostridium sensu stricto 1在对照组中含量较多。[Clostridium] innocuum group是一种革兰氏阳性、耐万古霉素、可形成孢子的共生微生物群成员,可导致抗生素相关性腹泻、严重结肠炎、肠外感染及克罗恩中的脂肪蠕动及肠道狭窄[12]。Ruminococcus gnavus是一种革兰氏阳性、非孢子形成的专性厌氧菌,其产生一种葡聚糖多糖,据推测这种多糖通过toll样受体4 (TLR4)诱导炎症反应,在胆石症的患者中富集,与胆汁酸代谢有关[13]。Mendez等[14]对痛风患者和健康对照组肠道菌群比较发现,健康对照组中Ruminococcus gnavus更富集,本研究结果与之不符,可能与本研究样本量较小有关,有待进一步扩大样本量研究具体情况。Shuttleworthia是属于梭菌属的厚壁菌门成员,它们是产生丁酸盐的专性厌氧菌,丁酸盐是结肠中不可消化碳水化合物发酵的主要产物[15]。本研究发现,HUA患者的Eisenbergiella数量增加。Eisenbergiella是从一名患有肥胖症的法国女性的粪便样本中分离出来的,还有证据[16]表明,专业健美运动员的肠道微生物组中富含该属的代表,这可能与他们的高蛋白饮食有关。Romboutsia是一种新的细菌,通过结肠镜检查从一名63岁的患有严重贫血和黑便症的法国男子的右人类结肠中分离出来,在一项中国的肥胖患者研究[17]中发现,Romboutsi、Clostridium为高尿酸、高血脂和高血压的肥胖患者常见的生物标志物。本研究中也发现HUA患者中Romboutsia丰度增加,Clostridium丰度下降。Clostridium sensu stricto可以产生丁酸盐,它会抑制全身炎症反应。Christensenellaceae R-7 group可以产生丁酸,它具有抗炎特性,可以下调促炎细胞因子,改善结肠黏膜屏障功能,同时丁酸是生产短链脂肪酸(SCFA)的重要底物。丁酸不仅是结肠上皮细胞的主要能量来源,还能抑制粘液中促炎细胞因子mRNA的表达[18]。我们发现HUA患者的Agathobacter的丰度降低,Agathobacter可通过膳食纤维的发酵产生SCFA,SCFA 通过抗炎作用对健康产生有益的影响,微生物群产生的SCFA产量减少会提高小鼠的腔内氧浓度,从而导致兼性厌氧菌的扩增[19]。
本研究也存在一些局限性。首先,我们可以评估此结果的样本相对较小,但提供了足够的统计能力来检测相关关联。其次,一项单中心研究可能会限制遗传和微生物相互作用的应用,未来的研究应扩展到在不同背景的多中心观察到的关联。第三,我们不是对整个DNA 进行鸟枪宏基因组测序,而是进行了16 SrDNA基因测序,这限制了物种水平和功能信息的数据解释,但大大拓宽了我们对与HUA相关的整体细菌结构和丰度的理解。而且,该研究的具体设计未能识别顺序因果关系,应在基因敲除动物模型或 HUA发病前后的纵向研究中对其进行调查。
总之,HUA发病和病情进展与肠道菌群失调和肠道微生物群的代谢功能改变有关。肠道微生物群中Romboutsia的富集与HUA相关,同时某些保护性菌群如Christensenellaceae R-7 group和Agathobacter丰度下降同样与HUA的发病可能有关联。由此可见肠道微生物群菌群的变化参与HUA疾病的发生与发展,具体机制有待进一步研究。
猜你喜欢
今日农业(2022年14期)2022-09-15
保健医苑(2022年1期)2022-08-30
中老年保健(2022年2期)2022-08-24
中国饲料(2022年5期)2022-04-26
昆明医科大学学报(2022年3期)2022-04-19
中老年保健(2021年4期)2021-08-22
中国医药科学(2021年5期)2021-05-11
中华养生保健(2020年4期)2020-11-16
保健与生活(2020年9期)2020-05-28
