
原肌球蛋白受体激酶抑制剂临床研究进展
2022-07-14 01:36刘淼刘晓韩开红韩开林
药学进展 2022年5期
刘淼,刘晓,韩开红,韩开林
(1. 济宁市精神病防治院内科,山东 济宁 272000;2. 天津市中央药业有限公司,天津 300400;3.济宁市精神病防治院精神科,山东济宁 272000;4.天士力控股集团有限公司研究院,天津 300410)
癌症是全球范围内威胁人类健康的重大疾病之一,在诊断和治疗方面,仅依靠肿瘤形态和免疫组学检查等传统手段来解决肿瘤的诊断和预后评估以及治疗管理等问题,已无法满足临床需求[1-2]。近年来,对大量特定的致癌突变(例如涉及关键致癌基因的基因激活点突变和染色体异常)的鉴定,将病理诊断转化为有效的治疗方案,即精准医疗,极大地改变了某些疾病的临床治疗方式,为医学技术发展开辟了新的道路。精准医疗广泛地应用于肿瘤研究和潜在有效药物靶标开发领域,促进了临床研究从“一刀切”向特定疾病人群的变革,有望解决临床需求[3-4]。
原肌球蛋白受体激酶抑制剂(tropomyosin receptor kinase inhibitor,TRKi)作为精准医疗的代表,是针对神经营养因子受体酪氨酸激酶(neurotrophin receptor tyrosine kinase,NTRK)融合基因的高选择性抑制剂,为携带NTRK融合基因的晚期实体瘤患者提供精准诊疗奠定了基础。作为针对特定基因突变而不限癌种的广谱抗癌药,虽然在临床上取得突破性疗效,但同样面临着获得性耐药的问题。因此,如何解决首代药物获得性耐药问题,将是未来该类药物研发的重点。本文通过对TRK抑制剂的治疗机制和研究进展进行综述,旨在为抗肿瘤的新药研发提供参考。
1 原肌球蛋白受体激酶
原肌球蛋白受体激酶A(tropomyosin receptor kinase A,TRKA)、TRKB和TRKC是受体酪氨酸激酶家族(receptor tyrosine kinases,RTK)重要成员之一,其分别由NTRK1、NTRK2和NTRK3编码。在神经系统正常发育过程中,TRK蛋白介导中枢和周围神经系统中神经元存活和突触可塑性[5-6]。NTRK1参与疼痛和体温调节;NTRK2参与运动、记忆、认知、情绪、食欲、体重调控;NTRK3在感知器官中发挥作用。TRKA、TRKB和TRKC是3种不同的跨膜蛋白,TRK蛋白由用于配体结合的细胞外结构域、跨膜蛋白结构域和具有酪氨酸激酶活性的胞内结构域组成[7-8]。其胞外域具有相似的结构和细胞内信号传导途径,但具有不同的激活和调节机制。这些受体以特定的生长因子作为配体,并参与细胞存活、生长、分化和凋亡等功能。当TRK功能失调导致下游信号通路被过度激活,从而诱导癌症的发生,其已成为潜在抗肿瘤药物靶点[9-12]。
1.1 原肌球蛋白受体激酶信号通路
TRK激酶结构域由1个的氨基末端和1个的羧基末端通过1个铰链区相连组成。与大多数酪氨酸激酶相似,受体与各自配体结合后导致受体的二聚化和自身磷酸化[13],随后激活丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)-细胞外调节蛋白激酶(extracellular signalregulated kinase,ERK)、磷脂酰肌醇3-激 酶(phosphoinositide 3-kinase,PI3K)-丝氨酸/苏氨酸激酶(protein kinase B,PKB,又称AKT)和磷脂酶C-γ(phospholipase C gamma,PLCγ)-蛋白激酶C(protein kinase C,PKC)等下游信号通路,从而调控细胞迁移、分化、突触形成和增殖等进程。正常情况下,神经生长因子(nerve growth factor,NGF)和TRKA受体的结合可促进神经元分化和轴突生长并介导过敏反应。脑源性神经营养因子(brain derived neurotrophic factor,BDNF)和神经营养因子4(neurotrophin-4,NT-4)与TRKB受体结合,调控中枢和外周神经系统,诱导神经细胞的增殖、分化和功能重塑。神经营养因子3(neurotrophin-3,NT-3)与TRKC受体结合介导PI3K-AKT信号途径,从而提高细胞存活率并避免细胞凋亡[14-17]。
当出现TRK融合蛋白时也可激活下游信号途径,如在结肠癌KM12细胞中,原肌球蛋白 3(tropomyosin 3,TPM3)-NTRK1嵌合蛋白的异常表达使得下游PI3K-AKT、Ras蛋白(ras proteins,RAS)-MAPK和PLCγ途径也异常活跃。TRK融合蛋白将处于持续活跃状态,引发永久性的信号级联反应,驱动TRK融合肿瘤的扩散和生长[18]。
1.2 神经营养因子受体酪氨酸激酶融合基因
NTRK融合基因发生在NTRK1、NTRK2或NTRK3与另一个不相关的基因之间,与不常见的致癌机制(NTRK突变、剪接变体和TRK过表达)相比,其代表具有致癌潜能的主要基因组改变。NTRK基因的致癌重排通常是由于NTRK基因的3′区和不相关基因的5′区融合引起的。NTRK融合基因保留了TRK受体的激酶结构域,5′区基因序列编码1个或多个可识别的二聚化结构域,TRK受体的激酶结构域与5′区不相关基因在框内融合形成新的融合蛋白[5,19]。TRK蛋白和NTRK融合基因机制如图1所示。
在不同类型的肿瘤中已报道了NTRK基因的突变,包括大肠癌、非小细胞肺癌(nonsmall-cell lung cancer,NSCLC)、黑色素瘤、急性粒性白血病、甲状腺癌、星型胶质细胞瘤、分泌性乳腺癌、脑胶质瘤、斯皮茨痣样黑素瘤、婴儿纤维肉瘤(infantile fibrosarcoma,IFS)、先天性中胚层肾癌等[20-25]。在所有实体瘤中NTRK融合蛋白的检出率约为1%,且不同肿瘤组织中其融合发生率存在较大差异。据基于肿瘤基因组图谱计划开发的集数据挖掘、整合和可视化等多功能于一体的综合性开放平台(cBioPortal)报道,在13%的肝癌、12%的浸润性乳腺癌、10%的肺腺癌和9%的子宫癌中报告了NTRK1拷贝数异常[26]。最新研究表明在10个乳腺癌的脑转移患者中有4个检出NTRK1融合基因,TRK介导的信号通路在转移性中枢神经系统疾病中发挥着关键调控作用[27]。此外,评估TRK mRNA和蛋白质表达水平的变化对一些恶性肿瘤的临床诊断和预后评估具有应用价值,研究显示在一些肾上腺癌、胰腺癌、卵巢癌、食道癌、膀胱癌和子宫内膜癌以及100%的嗜铬细胞瘤中均存在TRKA mRNA过表达和基因的扩增[28]。
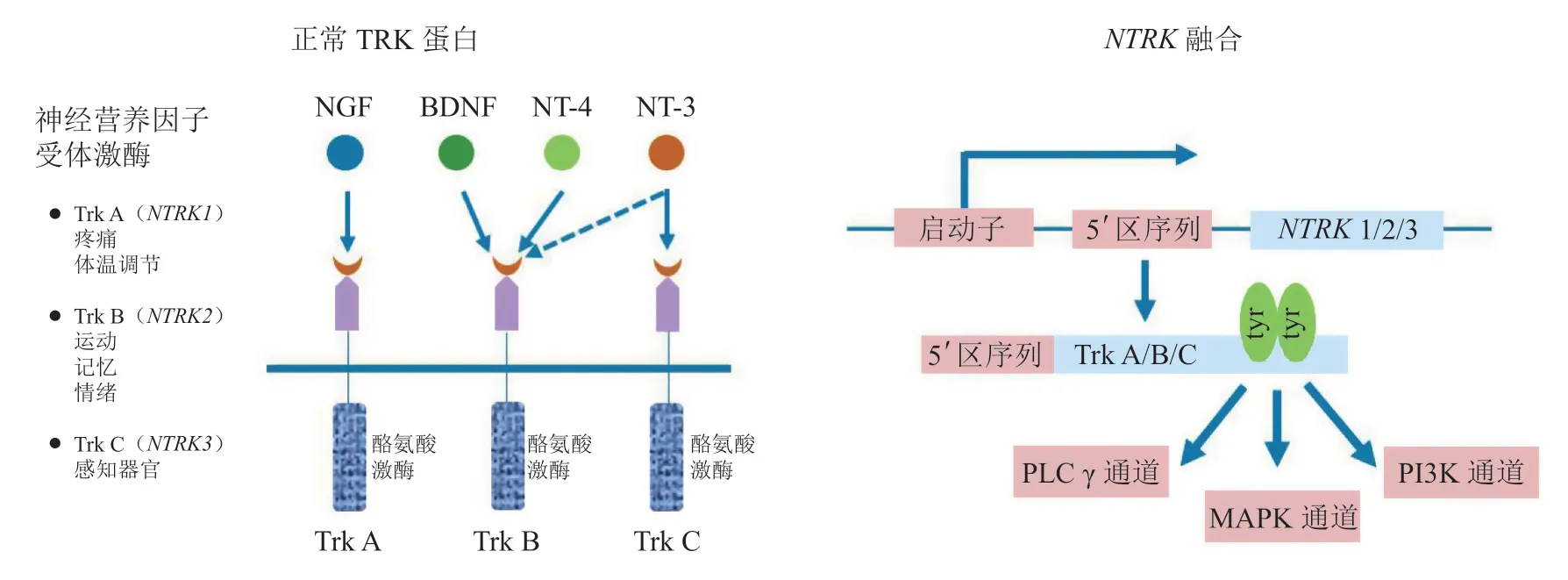
图 1 TRK蛋白和已知的NTRK融合基因产生机制[19]Figure 1 The normal TRK proteins and a schematic diagram of the known NTRK gene fusion
在NTRK基因的激活和催化环节中存在许多单核苷酸改变,但很少有致癌作用。例如,约5%的肺神经内分泌肿瘤与NTRK2和NTRK3基因的突变有关,其功能有待深入研究[29]。在成人转移性斯皮茨痣样黑素瘤中,NTRK1、NTRK2和NTRK3融合基因的检出率不足1%。在非小细胞肺癌中NTRK融合基因的发生率为0.1% ~ 3%,且呈现出NTRK融合与典型突变相斥的方式[30-31]。最近在1例复发/难治的转移性结直肠癌(colorectal cancer,CRC)患者中发现了一种新型核纤层蛋白A/C(recombinant lamin A/C,LMNA)-NTRK1融合蛋白,该患者使用恩曲替尼后达到部分缓解[32]。
鉴于NTRK融合基因作为关键的致癌驱动因子,促进癌细胞生长和存活,且可发生在人体任何部位。故以TRK蛋白为靶点的抗肿瘤药物的开发备受各个科研机构和制药公司的青睐。
2 原肌球蛋白受体激酶抑制剂
TRK抑制剂是通过阻断异常的TRK信号通路达到抑制增殖效果的小分子化合物,这些小分子能够在不同组织/不同肿瘤中起到良好的抑瘤效果。
2.1 原肌球蛋白受体激酶抑制剂分类及临床研究进展
近几年已开发出多种具有不同结构类型的TRK抑制剂,基于小分子抑制剂与TRK结合位点的差异,可将其分为3种类型,即Ⅰ、Ⅱ和Ⅲ型。靶向作用于腺苷三磷酸(adenosine triphosphate,ATP)结合位点的称为Ⅰ型激酶抑制剂;靶向作用于ATP结合位点和相邻疏水口袋的称为Ⅱ型激酶抑制剂;Ⅲ型激酶抑制剂也称为变构抑制剂。NTRK融合基因抑制剂大部分属于Ⅰ型激酶抑制剂,目前已上市的有拉罗替尼(1)、恩曲替尼(2),处于临床试验阶段有selitrectinib(3)、repotretinib(4)、AB-106(5)、HG030、BPI-28592、TQB-3558等;处于临床试验阶段的Ⅱ型TRK激酶抑制剂为altiratinib(6);处于临床试验阶段的Ⅲ型TRK激酶抑制剂为VMD-928(7)。
作为针对NTRK融合基因的广谱抗肿瘤药物,目前已有2个上市药物及多个TRK抑制剂处于临床研究阶段,表1总结了目前已上市及进入临床阶段的TRK抑制剂。
除已报道的进入临床阶段的TRK抑制剂外,越来越多的药物进入临床前研究,拟开发的适应证不仅局限于肿瘤领域,已向炎症、疼痛和神经系统等疾病领域拓展,表明研究者对生物标志物应用于药物研发和靶向药物递送方面所取得的重大进展,未来将会有更多的患者获得精准治疗。处于临床前阶段的TRK抑制剂见表2。

表 2 临床前研究阶段的原肌球蛋白受体激酶抑制剂Table 2 TRK inhibitors in preclinical trials
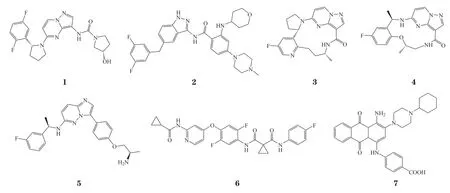

表 1 TRK抑制剂临床研究进展Table 1 Advances in clinical research on TRK inhibitor
下面将按研究阶段的不同,重点探讨临床阶段TRK抑制剂和近期报道的数据较优的处于临床前研究的TRK抑制剂(见表1、2),旨在为后续TRK抑制剂的开发及适应证选择提供依据。
2.2 已上市及处于临床阶段的原肌球蛋白受体激酶抑制剂研究进展
2.2.1 拉罗替尼拉罗替尼是拜耳和LOXO Oncology公司联合研发的Ⅰ类TRK抑制剂,用于治疗携带NTRK融合基因且不可手术或转移性实体瘤,2018年11月26日在美国获批上市。
体外研究发现拉罗替尼是一种高选择性、口服给药的ATP竞争性抑制剂,其对TRKA、TRKB和TRKC的IC50分别为6.5、8.1和10.2 nmol · L-1,对带有NTRK融合基因的细胞较为敏感,而对其他激酶无抑制活性[33]。临床前体内/外测试结果表明,拉罗替尼对携带NTRK融合基因的抑癌效果显著。在KM12异种移植瘤裸鼠模型中,灌胃给药2周,肿瘤体积明显减小。多种TRK融合肿瘤模型试验结果表明,拉罗替尼通过靶向抑制MAPK、PKC、PI3K/AKT和信号传导转录激活因子3(signal transducer and activator of transcription 3,STAT3)信号通路,达到抑制TRK融合肿瘤的效果[34]。
在一项剂量递增的Ⅰ期临床试验(NCT02122913)中,70例肿瘤患者入组,口服给药,28 d为1个周期,在给药剂量未达到最大耐受剂量(maximum tolerated dose,MTD)时即显示出良好的安全性和抑瘤效果,建议给药剂量为每次100 mg。其中,62例患者NTRK融合基因阴性,8例患者带有NTRK融合基因[6个转录因子ETS差异基因6(ETS variant gene 6,ETV6)-NTRK3、1个LMNANTRK1、1个转录辅抑制因子(topless related,TPR)-NTRK1],在NTRK融合基因患者中,经研究者评估的客观缓解率(objective response rate,ORR)为88%,中位随访时间分别为26.9和27个月,未达到缓解持续时间(duration of response,DOR)[35]。
Ⅰ/Ⅱ期临床试验(NCT02637687、NCT02576431、NCT02122913)结果显示,接受拉罗替尼治疗后患者ORR为79%,其中24%的患者达到完全缓解(complete remission,CR)和53%的患者部分缓解(partial response,PR),其在所有NTRK融合基因的肿瘤中均具有显著的抑瘤活性,且抑瘤活性不受患者年龄和肿瘤类型的影响。在中位随访8.3个月时,未达到中位缓解持续时间,并且在1年时间内,约有71%的患者出现了缓解,显示出持久的抗肿瘤活性[36-37]。2022年4月8日,拉罗替尼获得中国国家药品监督管理局(National Medical Products Administration,NMPA)批准,由拜耳医药保健有限公司销售。
2.2.2 恩曲替尼恩曲替尼是罗氏和Ignyta Pharma公司联合开发新型、口服酪氨酸激酶抑制剂,用于治疗携带NTRK1/2/3、c-ros肉瘤致癌因子-受体酪氨酸激酶(ros proto-oncogene 1,ROS1)基因和ALK融合基因的晚期或复发的实体瘤,这些基因融合出现在约40种原发实体瘤中,于2019年在日本和美国上市[38]。
恩曲替尼是5种靶标致癌驱动因子(TrkA、TrkB、TrkC、ROS1和ALK)高选择性的ATP竞争抑制剂,其IC50分别为1、3、5、7和12 nmol · L-1[39]。与克唑替尼(经批准治疗ROS1融合阳性NSCLC)相比,恩曲替尼对ROS1的效力较其高30倍[40]。
Ⅰ期临床试验结果显示,恩曲替尼在16例携带NTRK、ROS1、ALK融合基因突变阳性的颅外疾病患者(n= 24)中ORR高达79%,并且在中枢神经系统(central nervous system,CNS)疾病的患者中也实现了完全的、可持续的缓解。在Ⅱ期STARTRK-2试验(NCT02568267)中,NTRK融合阳性实体瘤患者接受恩曲替尼600 mg(qd)剂量的治疗,56.9%达到了ORR,基线CNS转移患者的颅内ORR为54.5%,其中27.3%的患者获得了CR,颅内缓解持续时间无法评估;CNS转移患者中,颅内ORR为50%,DOR为10.4个月[41-42]。
恩曲替尼具有良好的耐受性,大多数与治疗有关的不良事件(n= 355)为轻度至中度(即1或2级)且可逆(Ⅰ/Ⅱ期试验的综合分析),在综合分析中没有报告5级治疗相关的不良事件。通过减少剂量(27%)或中断(25%)来控制与治疗相关的不良事件;4%的患者需要停药,8.5%的患者发生了严重的不良事件[40,42-43]。2019年在日本和美国上市,国内处于上市申请阶段。
2.2.3 SelitrectinibSelitrectinib(LOXO-195)是由拜耳和LOXO Oncology公司研发的口服、高效、高选择性的第2代TRK 抑制剂,selitrectinib的临床前研究与拉罗替尼早期临床研究同步进行,旨在解决NTRK融合基因阳性患者耐药性问题。
Selitrectinib在体外能有效地抑制不同的活化TRK激酶,其针对野生型TRKA(WT)、突变型TRKA(G667CA)、TRKA(G595R) 和TTRKA(F589L)的IC50分别为0.35、16.7、5.7和10.9 nmol · L-1。Selitrectinib对TRK激酶具有高选择性,在浓度为1 μmol · L-1时,对ROS激酶的抑制率达到88%,对活化的Cdc42相关激酶1(activated Cdc42-associated tyrosine kinase 1,ACK1)的抑制率达到90%,对TXK酪氨酸激酶(txk tyrosine kinase,TXK)的抑制率达到86%。在TRK融合突变的KM12、Ba/F3 LMNA-NTRK1(F589L)、Ba/F3ETV6-NTRK3(G623R)和O-91细胞系中,selitrectinib能有效地抑制其增殖(IC50≤5 nmol · L-1)。而在不包含TRK融合的84种细胞中,LOXO-195浓度即使高达10 μmol · L-1对细胞生长也无抑制作用[44]。
Ⅰ/Ⅱ期临床方面:Nature Review杂志报道了31例NTRK融合基因耐药患者接受selitrectinib治疗的相关数据(其中20例患者来自selitrectinib的Ⅰ期临床试验,11例患者来自同情用药计划,这些患者前一次NTRK靶向药治疗中位持续时间为9.5个月),易发生溶剂前沿突变(如TRKAG595R和TRKCG623R)的患者ORR可达到45%。
2.2.4 RepotrectinibRepotrectinib(TPX-005)是 由Turing Point Therapeutics公司研发的多靶点抑制剂,用于治疗携带ALK、ROS1或NTRK致癌基因重排的非小细胞肺腺癌,研究表明repotrectinib克服了ROS1、NTRK1/2/3和ALK的获得性溶剂前沿突变而引起的耐药性问题[45-47]。
Repotrectinib对ROS1(WT)、TRKA、TRKB、TRKC和ALK的IC50为0.071 ~ 4.46 nmol · L-1。在100 nmol · L-1浓度下,进行了针对395种激酶选择性的筛选,其中对ROS1(WT)、TRKA、TRKB、TRKC和ALK融合基因突变的细胞具有高选择性。通过对小鼠异种移植瘤模型进行体内药效研究,在携 带CD74-ROS1/BaF3(WT)或CD74-ROS1G2032R异种移植瘤的SCID/Beige小鼠中,每天2次,给药剂量为15或75 mg · kg-1,针对CD74-ROS1(WT)小鼠的肿瘤生长抑制(target group index,TGI)分别为197%和200%,CD74-ROS1G2032R的TGI分别为99%和200%[48-50]。
Ⅰ期临床数据在2019年美国临床肿瘤学会(American Society of Clinical Oncology,ASCO)大会上公布,入组33例ROS1融合基因突变患者(11例未接受任何靶向药物的治疗,22例接受过ROS1靶向药物的治疗),在未接受过靶向药物治疗的患者中repotrectinib的ORR为82%,其中3例脑转移患者的脑部病灶全部客观缓解,持续缓解时间均达到10个月以上;在接受过ROS1靶向药物的治疗的患者中repotrectinib的ORR为39%,4例脑转移患者中2例脑部病灶客观缓解,显示出repotrectinib对脑转移具有很好的控制作用。在安全性方面repotrectinib的不良反应为1 ~ 2级,相较于其他药物安全性较好[51]。2020年7月,再鼎医药与Turning Point Therapeutics公司签署一项独家授权协议,推进repotrectinib在中国的开发及商业化。目前国内处于Ⅲ期临床研究中。
2.2.5 AB-106AB-106是第一三共制药和葆元生物医药共同研发的口服小分子ROS1/NTRK抑制剂,用于治疗携带NTRK融合基因的实体瘤和ROS1融合阳性的局部晚期或转移性非小细胞肺癌。体外研究发现AB-106是一种高选择性、口服给药的ATP竞争性抑制剂,其针对ROS1、NTRK1、NTRK2、NTRK3的IC50分 别 为0.21、0.62、2.28、0.98 nmol · L-1,在 小鼠异种移植瘤模型中,口服给药剂量25 mg · kg-1时,针对含有原肌球蛋白3(tropomyosin3,TPM3)-NTRK1融合基因的KM12细胞和具有FIG-ROS1融合基因的U-118MG细胞的生长被显著抑制。在Ba/F3-ROS1异种移植瘤小鼠中,也观察到肿瘤的消退,而克唑替尼没有明显作用[52]。
在一项安全性、耐受性和药效评估的Ⅰ期临床中,入组15例患者(均为ROS1融合阳性的NSCLC患者),剂量递增结果显示,基于AB-106暴露量的差异,在日本,AB-106的耐受剂量和Ⅱ期推荐剂量为600 mg,美国的最大耐受剂量为800 mg,主要不良反应事件为2级,80%患者中观察到丙氨酸转氨酶(alanine aminotransferase,ALT)和天冬氨酸转氨酶(aspartate aminotransferase,AST)升高,主要不良事件为腹泻、恶心,整体耐受性良好。在未曾使用克唑替尼治疗的ROS1融合的NSCLC患者中,ORR为66.7%,DCR为100.0%,这些结果与克唑替尼(ORR为72%)相似,AB-106针对ROS1融合突变的NSCLC患者具有较好的抗肿瘤效果,由于样本量较少,有待在临床中进一步验证[53-55]。
2.2.6 PLX-7486PLX-7486是由Plexxikon公司研发的口服小分子TRK激酶抑制剂,用于治疗具有神经、炎症特征的实体瘤。
PLX-7486临床前数据已在第102届美国癌症研究学会(American Association for Cancer Research,AACR)会议上公布,其具有良好的口服生物利用度,在含有BAF3 TRK和BAF3 FMS细胞的小鼠异种移植瘤模型中,PLX-7486能够很好地抑制肿瘤的生长,PLX-7486对骨髓来源的巨噬细胞RAW264.7还显示出较强的抑制作用(IC50<1 μmol · L-1)。研究人员在MC38和B16F10小鼠肿瘤模型中研究了PLX-7486与免疫抑制剂(例如抗CTLA-4和PD-1)联合用药的抗肿瘤效果,结果显示联合用药组的抑瘤效果明显优于单药组[56]。
PLX-7486Ⅰ期临床试验已完成针对晚期实体瘤患者的安全性、药动学和药效学试验(NCT01804530),结果尚未公布。临床前组合用药已有初步疗效,为临床上组合用药方案提供了研究基础,PLX-7486与吉西他滨组合用药旨在获得安全的组合剂量来达到比单独用药更优的疗效,由于Ⅱ期试验结果未达到预期而终止,现处于终止状态[57]。
2.2.7 VC-004VC-004是由江苏威凯尔医药科技有限公司自主研发的口服小分子第2代TRK激酶抑制剂,2020年11月20日,在中国开展Ⅱ期临床试验(NCT04614740、NCT02094586),用于治疗实体瘤,预计2024年1月完成患者入组。
2.2.8 ICP-723ICP-723是由诺诚健华研发的口服第2代泛TRK小分子抑制剂,用于治疗携带NTRK融合基因的晚期或转移性实体瘤及第1代TRK抑制剂治疗获得性耐药的患者。Ⅰ期剂量递增(1、2、3和4 mg)研究显示(NCT04685226),在2名NTRK融合患者中显示出疗效,3 mg队列中的NTRK融合阳性患者达到疾病稳定,在4 mg队列中的患者在第1周期结束时实现了部分缓解。目前,旨在评估安全性、耐受性和药动学特性的Ⅰ/Ⅱ期临床试验正在招募中。
2.2.9 FCN-011FCN-011是复星医药全资子公司重庆复创医药研发的口服小分子TRK抑制剂,拟用于治疗NTRK融合基因阳性的实体瘤,2021年4月由重庆复创医药研究有限公司在中国开展Ⅱ期临床试验(NCT04687423),用于治疗实体瘤,2021年4月13日在国内完成首例受试者入组,预计2023年10月31日完成招募。
2.2.10 PBI-200PBI-200是Pyramid Biosciences Inc公司开发的口服高透血脑屏障的第2代TRK抑制剂,2021年7月20日,在美国启动Ⅱ期临床试验(NCT04901806),用于治疗促结缔组织增生性小圆细胞肿瘤、脑癌和实体瘤,预计2024年6月完成患者招募。
2.2.11 BPI-28592BPI-28592是贝达药业研发的第2代口服TRK激酶抑制剂,广谱抗肿瘤药,用于治疗携带NTRK基因融合的局部晚期或转移性实体瘤。在2020年6月AACR年会上公布的临床前结果显示,在1 000 nmol · L-1浓度下BPI-28592针对468种激酶具有显著的选择性,其中对TRKA、TRKB、TRKC、促红细胞生成素产生肝细胞B6
(erythropoietin-pro-ducing hepatocyte B6,EPHB6)、
ROS1和TNK2的结合常数分别为0.23、0.55、1.1、30、19和100 nmol · L-1。在原代和耐药TRK突变细胞的异种移植瘤小鼠模型中,每天1次或2次给药剂量1.5 ~ 200 mg · kg-1,BPI-28592对肿瘤的抑制程度呈明显的药物剂量依赖性。该药于2020年4月获得NMPA临床默示许可,目前正进行患者招募(NCT05302843),预计2023年12月31日完成患者招募。
2.2.12 TL118TL118是由苏州韬略生物科技有限公司自主研发的“不限癌种”的NTRK靶向药,旨在解决第1代TRK激酶入脑能力差、耐药性和选择性不高等问题,用于治疗携带NTRK基因融合的晚期恶性实体瘤。2019年8月获得NMPA临床试验默示许可,目前正进行患者招募,相关结果尚未公布。TQB-3558是由正大天晴研发的口服小分子TRK激酶抑制剂,用于治疗晚期实体瘤,2020年4月该药获得NMPA临床试验默示许可,Ⅰ期临床研究正在进行中(NCT04408079),预计2022年7月完成患者招募。
2.2.13 HG030HG030是由成都先导药物开发股份有限公司开发的高活性、高选择性的第2代口服小分子ROS1/TRK双靶点抑制剂,用于携带NTRK及ROS1融合基因的局部晚期或转移性实体瘤患者的治疗。2019年6月公布的临床前数据显示,HG-030对野生型和突变型TRK激酶具有显著的抑制作用(IC50<1 nmol · L-1)。在小鼠胚胎成纤维细胞3T3 TRKA(G595R)小鼠移植瘤(cell line derived xenografts,CDX)动物模型中,HG-030(给药剂量10 mg · kg-1)具有显著的抑瘤作用,抑制率达98%。在小鼠中HG-030显示出良好的口服生物利用度(27.7%),半衰期为1.9 h。该药2020年3月获得NMPA临床试验默示许可,目前正进行Ⅰ期临床病例招募,尚未完成首例受试者入组。2020年11月10日,广州白云山医药集团股份有限公司获得HG030在中国后续研发、生产及销售权益。2022年1月获得美国FDA临床试验默示许可。
2.2.14 AltiratinibAltiratinib(DCC-2701)是Deciphera公司研发的口服小分子多靶点激酶抑制剂,用于实体瘤患者的治疗。其针对TRK1、TRK2、TRK3、MET、内皮细胞TEK酪氨酸激酶(recombinant TEK tyrosine kinase,endothelial,TIE2)、血管内皮细胞生长因子受体2(vascular endothelial growth factor receptor 2,VEGFR2)和FMS样酪氨酸激酶3(FMS-like tyrosine kinease 3,FLT3)的IC50分 别为0.85、4.6、0.83、2.7、8、9.2和9.3 nmol · L-1,altiratinib在野生型和突变型NTRK融合突变的细胞中能显著诱导肿瘤细胞的凋亡,altiratinib可以穿越血脑屏障,具有治疗脑癌和脑转移癌的潜力。在小鼠胶质母细胞瘤异种移植瘤模型中,与贝伐单抗联用比单独用药可有效延长生存期。用于治疗局部晚期肿瘤和转移性实体瘤的Ⅰ期临床研究已完成,但结果尚未公布[58-59]。
2.2.15 VMD-928VMD-928是由VM Oncology LLC公司开发的口服小分子TRKA拮抗剂,目前正进行治疗实体瘤和淋巴瘤的Ⅰ期临床试验(NCT03556228),预计2023年12月完成患者招募[60]。
2.2.16 LY01018LY01018是由绿叶制药研发的高选择性、口服第2代NTRK小分子抑制剂,其对野生型和获得耐药型突变均有效,用于治疗NTRK基因融合及其耐药突变阳性的实体瘤,2020年8月获得NMPA临床默示许可。
2.2.17 FCN-098FCN-098是重庆复创医药研究有限公司自主研发的口服第2代TRK抑制剂,用于治疗NTRK基因融合的晚期实体瘤,2021年6月获得NMPA临床默示许可,2022年1月已启动旨在评估晚期在实体瘤患者中的安全性、耐受性、药动学特征和原发性抗肿瘤活性Ⅰ期临床试验(NCT05212987),预计2024年5月完成患者招募。
2.2.18 SIM-1803-1ASIM-1803-1A是先声药业自主研发的新1代选择性NTRK/ROS1多靶点酪氨酸激酶抑制剂,用于治疗NTRK/ROS1基因融合的晚期或转移性实体瘤,2021年1月在中国完成Ⅰ期临床试验首例受试者给药(NCT04671849),临床正在进行中,预计2022年9月完成患者招募。
2.2.19 HL-5101HL-5101是由韩国Handok Inc和Cmg Pharmaceutical公司共同研发的一种高选择性泛TRK抑制剂,于2019年08月在韩国启动Ⅰ期临床试验(NCT04014257),用于治疗实体瘤,2021年9月完成全部17例受试者入组,结果尚未公布。
2.3 原肌球蛋白受体激酶抑制剂治疗炎症、疼痛和神经系统等疾病临床研究进展
目前TRK抑制剂是基于生物标志物而非肿瘤位置进行区分的“精准疗法”,在抗癌药物的研发历程上具有里程碑式的意义。但其在炎症、疼痛和神经系统疾病领域的临床研究结果尚未达到预期目标,还存在许多亟待解决的的问题。
AK-1830是由辉瑞子公司Array BioPharma Inc研发的口服小分子TRKA抑制剂,拟开发的适应证为炎症、关节炎和疼痛。2019年10月在日本启动了针对疼痛、炎症的Ⅰ期临床试验,同时在2020年1月进行了治疗关节炎伴疼痛的Ⅱ期临床试验(JapicCTI-195073)。临床正在进行中,相关数据尚未公布。
PBI-100是阿斯利康和Pyramid Biosciences共同研发的外用小分子NTRK调节剂,拟开发适应证为皮肤性疾病、银屑病和特应性皮炎等。2021年4月在美国启动评估PBI-100外用乳膏对健康受试者的皮肤刺激的Ⅰ期临床研究(NCT04882631),2021年5月完成30例受试者入组,结果待公布。
ASP-7962是安斯泰来制药有限公司研发的新型口服、选择性小分子TRKA抑制剂,用于骨关节炎(osteoarthritis,OA)患者的治疗。体外研究发现,ASP-7962抑制ATP诱导的TRKA、TRKB和TRKC激酶的磷酸化,其IC50分别为0.155、1.41和1.09 μmol · L-1,临床前模型、毒性研究及Ⅰ期临床剂量递增试验均支持选择100 mg(bid)人体剂量作为最佳治疗剂量。评估ASP-7962的安全性和耐受性的Ⅰ期临床研究(NCT01981928和NCT02136316)表明,健康受试者中单次剂量递增至240 mg和多次剂量200 mg(bid)的安全性良好。Ⅱa期临床试验中215例OA患者被随机分组(ASP-7692n= 85;安慰剂n= 87;萘普生n= 43)服用ASP-7962 100 mg(bid)、安慰剂或萘普生500 mg(bid),持续给药4周。结果显示,在任何西大略和麦克马斯特骨关节炎指数(Western Ontario and McMaster Universities Osteoarthritis,WOMAC)评分量表分值中,未观察到ASP-7962和安慰剂之间的差异,萘普生和安慰剂之间观察到统计学上显著变化(P≤0.01)。因此,使用ASP-7962 100 mg(bid)进行4周治疗并未改善OA患者的疼痛,导致项目终止[61]。
GZ-389988是由赛诺菲从Genzyme公司收购的小分子TRKA抑制剂,用于OA的治疗。临床前数据显示,在碘乙酸钠诱导的OA和链球菌肽聚糖多糖介导的关节炎大鼠模型中,单剂量持续给药4周降低了局部关节炎症反应,且耐受性良好。Ⅰ期临床试验评估了GZ-389988单次递增的OA剂量的疗效、安全性、耐受性和药动学性质,于2016年7月完成,结果尚未公布。Ⅱ期临床试验针对OA患者评估了GZ-389988疗效、安全性、耐受性和药动学特性,数据未公布。近几年赛诺菲因为加大向肿瘤和特定疾病特效药领域聚焦,因此GZ-389988项目被迫终止。
2.4 原肌球蛋白受体激酶抑制剂耐药机制
随着第1代TRK抑制剂拉罗替尼和恩曲替尼在临床上的应用,获得性耐药问题已出现。从目前报道的数据上看,获得性耐药问题主要发生于TRK激酶结构域中点突变或旁路途径突变位点,造成针对这些突变的NTRK融合癌的治疗效果较差。在一项对拉罗替尼获得性耐药研究中发现,突变体的TRK蛋白与正常蛋白相比,增强了与ATP的亲和力,从而阻止了拉罗替尼与激酶的结合。针对恩曲替尼获得性耐药研究发现,其主要发生于NTRK1和NTRK3激酶域。涉及的耐药突变包括:
NTRK1G595R、NTRK1F589L、NTRK1G667C、NTRK2G639R、NTRK3G623R、NTRK3G696A和NTRK3F617L[62-63]。是否有其他因素参与了耐药有待后续研究的进一步探索。
基于现阶段TRK抑制剂的获得性耐药问题,从产生耐药的分子机制入手,进行全新第2代TRK抑制剂的开发,有望克服第1代耐药性问题。目前已有LOXO-195、repotrectinib、BPI-28592、TL118、ICP-723和HG030等多个药物进入临床研究阶段。
3 结语与展望
NTRK融合蛋白在多种成人和儿童癌症的发生发展中扮演了重要的角色,小分子TRK抑制剂用于NTRK融合蛋白癌症的治疗,具有广谱抗肿瘤作用,且副作用较小。随着更准确、更高效的检测技术的应用,例如荧光原位杂交(fluorescence in situ hybridization,FISH)、肿瘤组织的下一代测序(next-generation sequencing,NGS)和循环肿瘤细胞(circulating tumor cell,CTC)测序等技术,将提供更精准的方法来检测NTRK融合蛋白,为临床上新的治疗策略指明了方向。
目前,TRK抑制剂更多地聚焦于肿瘤领域,为避免同质化竞争问题,适应证的拓展势在必行,已报道多个小分子TRK抑制剂用于炎症、疼痛和神经系统疾病的治疗,且有4个处于临床研究阶段,6个处于临床前研究阶段。但令人遗憾的是,针对炎症、疼痛和神经系统疾病的临床疗效未达到预期目标。其中,ASP-7962治疗骨关节炎的Ⅱ期临床试验以失败告终;GZ-389988因赛诺菲研发管线收缩而终止。这并不代表其在炎症、疼痛和神经系统疾病治疗方面的失败,而揭示了TRK抑制剂在炎症、神经系统疾病等适应证开发上还存在许多亟待解决的问题。
第1代TRK抑制剂拉罗替尼和恩曲替尼已被批准用于治疗携带NTRK融合蛋白的儿童和成人转移或局部晚期的实体瘤,临床上取得了卓越的抗癌效果。但也面临着TRK抑制剂治疗的获得性耐药问题,目前正进行基于序列、结构和动力学方法进行新一代TRK抑制剂研发和针对TRK细胞内信号传导途径及耐药机制的探索,相信耐药性问题将很快得到解决。
猜你喜欢
中国典型病例大全(2022年11期)2022-05-13
健康体检与管理(2022年4期)2022-05-13
医学概论(2022年4期)2022-04-24
中国药房(2022年7期)2022-04-14
海外星云(2021年9期)2021-10-14
现代仪器与医疗(2021年1期)2021-06-09
大众健康(2020年7期)2020-08-25
爱你(2019年13期)2019-11-14
科学24小时(2018年1期)2018-01-10
现代养生·下半月(2016年6期)2016-10-21
