
选择性酪氨酸激酶2抑制剂的研究进展
2022-07-14 01:36王力勋朱佩钰周金培张惠斌
药学进展 2022年5期
王力勋,朱佩钰,周金培,张惠斌
(1.中国药科大学新药研究中心,江苏 南京 210009;2.中国药科大学药物化学教研室,江苏 南京 210009)
细胞因子是调节机体自身免疫的关键因子,例如白细胞介素(interleukin,IL)、干扰素(interferon,IFN)等细胞因子都通过Janus激酶(Janus kinase,JAK)信号转导和转录激活因子(signal transducers and activators of transcription,STAT)途径传递信号[1]。当细胞因子异常表达或细胞内通路失调时,免疫稳态被破坏,引起慢性炎症、自身免疫性疾病以及肿瘤的发生。抑制JAK可以阻断JAK-STAT通路,使介导人类疾病的相关传递受阻,从而阻断疾病的发生与发展[2]。这一策略成为治疗类风湿关节炎、银屑病和炎症性肠病(如克罗恩病、溃疡性结肠炎)等自身免疫性疾病的选择[3-4]。JAK家族共有4位成员,包括JAK1、JAK2、JAK3和酪氨酸激酶2(tyrosine kinase,TYK2)。其中,TYK2参与IL-12、IL-23和Ⅰ型IFN的信号传导,且IL-12和IL-23的异常表达会引起银屑病和炎症性肠病等自身免疫性疾病[5-7]。研究发现,选择性抑制TYK2能够在临床治疗中避免广泛的免疫抑制[8],已成为治疗自身免疫性疾病的潜在策略[9-10]。本文通过对TYK2的生物学结构、通路作用机制及其抑制剂的研究进展进行综述,以期为代谢性疾病药物的发展研究提供参考。
1 酪氨酸激酶2的生物学功能
1.1 Janus激酶家族及其功能
JAK家族成员,包括JAK1、JAK2、JAK3和TYK2,是细胞因子受体相关的非受体酪氨酸蛋白激酶,其在结构上具有高度同源性,由7个不同结构域组成,从C端到N端分别为JH1 ~ JH7。根据JAK家族结构域的功能差异,7个不同结构域可分成4个不同功能域:1)激酶结构域(JH1),是腺嘌呤核苷三磷酸(adenosine triphosphate,ATP)活性位点所在结构域,负责底物磷酸化;2)假激酶结构域(JH2),与JH1结构相似,但缺乏催化功能,参与调控JH1催化活性;3)Src同源2结构域(Src homology domain2,SH2)(JH3与JH4),用以稳定 JAK构 象;4)FERM(band-four-point-one ezrin radixin moesin)同源结构域[11](见图1)。
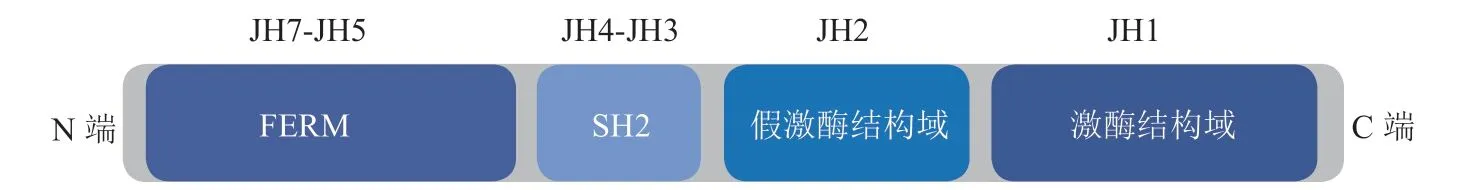
图 1 JAK家族结构域Figure 1 Domain of Janus kinases
JAK家族蛋白在哺乳动物生命活动中起着重要作用。其中,JAK1缺失导致小鼠淋巴细胞增殖严重受损;JAK2能诱导并调节红细胞生成,当JAK2缺失时导致血小板减少,其过表达则与骨髓增生性疾病相关,如真红细胞增多症和原发性血小板增多症[12];JAK3主要在淋巴组织和造血组织中表达,JAK3缺失的小鼠表现出严重的联合免疫缺陷症状(severe combined immunodeficiency,SCID)[13];
T
YK2则在免疫中发挥关键作用,缺乏TYK2的小鼠可以正常发育,然而其感染分枝杆菌或病毒的风险增加, TYK2缺失的患者则表现出高IgE综合征(hyper-IgE syndrome,HIES)[14]。
1.2 Janus激酶-转录激活因子信号通路
JAK-STAT信号通路是细胞因子信号传导的重要途径,参与人体内细胞增殖、分化、凋亡及免疫调节等过程。TYK2是JAK-STAT通路中的一种关键信号转导激酶,在传递炎症因子和免疫应答信号方面具有重要的作用。如图2所示,TYK2与JAK2共同作用于IL-12和IL-23信号通路,或者与JAK1共同作用调节Ⅰ型IFN的传导[1]。IFN或IL与受体特异性结合后导致受体二聚化,随后招募JAK蛋白向受体细胞质聚集并磷酸化激活JAK。与此同时活化的JAK磷酸化STAT蛋白,STAT形成二聚体并转运至细胞核内,与目标基因启动子结合后调节特定基因的表达[15]。JAK抑制剂能够与JAK蛋白的ATP位点结合并抑制JAK的磷酸化激活,由此破坏下游的级联反应,导致自身免疫性疾病、炎症等疾病相关基因转录减少[16]。
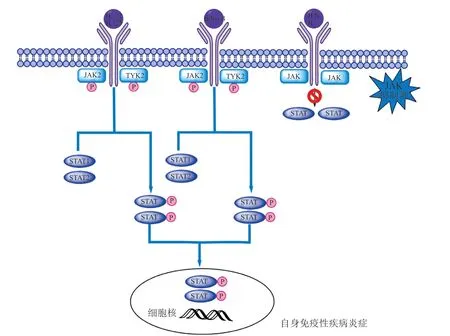
图 2 JAK-STAT信号通路和JAK抑制剂作用机理Figure 2 JAK-STAT signaling pathway and mechanism of JAK inhibitor
1.3 酪氨酸激酶2与自身免疫性疾病
研究表明,人体生命活动中需要TYK2参与的通路,仅限于IL-12和IL-23介导的途径,IL-6或IL-10的信号传导则不需要TYK2参与,因此抑制TYK2可能是治疗免疫性疾病的有效方法[8]。值得注意的是,在高剂量脂多糖诱导的内毒素休克、肠缺血/再灌注休克、胶原诱导的关节炎、实验性自身免疫性脑脊髓炎及过敏性气道炎症等模型研究中,TYK2的缺失都具有保护作用,可以增加机体对自身免疫性疾病、过敏和炎症性疾病的抵抗力[17]。全基因组关联研究表明,一种失活的TYK2突变体对包括多发性硬化症、炎症性肠病、强直性脊柱炎和银屑病在内的多种免疫性疾病具有治疗保护作用[18]。多种抗体的临床疗效证明了抑制TYK2信号通路在自身免疫性疾病治疗中的价值:1)靶向IL-12和IL-23信号通路的ustekinumab在临床上已被批准治疗银屑病、关节炎和克罗恩病[19];2)近期临床研究表明,ustekinumab对系统性红斑狼疮患者显示出疗效[20];3)能够特异性阻断IL-23信号传导的guselkumab可用于银屑病的临床治疗[21]。因此,TYK2是自身免疫性疾病领域的重要靶点,选择性TYK2抑制剂能取得更安全有效的临床疗效。
2 选择性酪氨酸激酶2抑制剂研究进展
鉴于TYK2在自身免疫疾病领域的突出作用,针对靶向TYK2的选择性抑制剂的研究一直在火热进行中。由于TYK2和JAK家族其他成员的激酶结构域具有高度同源性[22],该研究充满挑战性。迄今为止,批准上市的JAK家族抑制剂主要与JH1的ATP位点结合,通过阻断ATP来抑制激酶的催化活性,从而干扰相关细胞因子信号转导,发挥治疗作用[23]。研究发现在JAK家族的 JH1 ATP位点中,有一个共同的守门氨基酸——蛋氨酸。JAK家族成员区别在于,TYK2的门控残基为异亮氨酸(Ile960),其他3个成员的门控残基是缬氨酸(Valine)[24]。这种独特的氨基酸差异为高选择性TYK2抑制剂基于结构的设计提供了依据。近年来,科学家在TYK2假激酶结构域的蛋白质结构和分子机制方面取得了重大进展,揭示了靶向TYK2 JH2的小分子可以调节相邻JH1结构域的构象,从而影响JH1的激酶活性[25-26]。研究表明,与JH2结合的小分子可以稳定JH2结构域和JH1 ATP活性位点之间的自抑制作用。而这些JH2-JH1相互作用阻碍了JH1活性位点催化所必需的氨基酸残基Tyr1054和Tyr1055的磷酸化。与TYK2 JH2结合的变构抑制剂可以在实现最大功效的同时保持较高选择性。本文将选择性TYK2抑制剂按照结构分类进行综述。
2.1 单环类
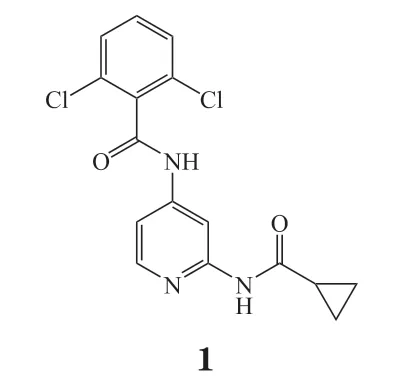
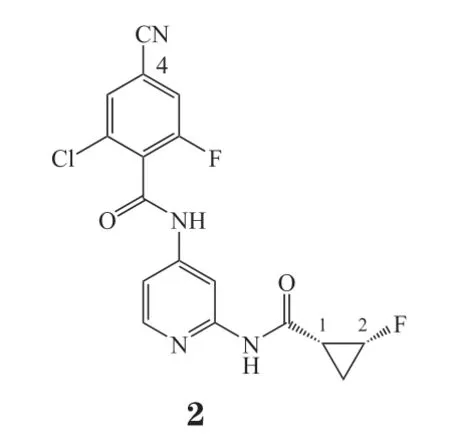
在2010年,罗氏公司开发了含有4-氨基吡啶苯甲酰胺的化合物1和化合物2作为口服选择性酪氨酸激酶2抑制剂,用于治疗银屑病和炎症性肠病[27]。通过高通量筛选得到化合物1(IL-12-pSTAT4 EC50=380 nmol · L-1),将其作为先导化合物,进行结构优化工作。在化合物1与JAK2和TYK2酶的JH1结合晶体结构中,2种酶的P-loop构象存在显著差异。与前者相比,TYK2的P-loop朝向C末端叶偏移,而化合物1结构中的苯环4位距离P-loop尖端的Glu905骨架羰基只有3.6×10-10m。因此,在化合物1的苯环4位引入氢键供体取代基可以与Glu905羰基产生作用,并可以与含有带电残基的极性区域(如Arg1027)相互作用。环丙酰胺基团与JH1结合处,JAK2与TYK2也存在残基差异,分别为谷氨酰胺(Gln853)和精氨酸(Arg901)。研究发现环丙酰胺水解和2,6-二氯苯环氧化是2种代谢途径。根据上述结论进行改造,苯环4位筛选得到最佳取代基——氰基(对TYK2的Ki为1.8 nmol · L-1),优于化合物1(Ki= 4.8 nmol · L-1)。而在环丙基引入顺式C2氟取代可以显著提高活性 (Ki= 2.5 nmol · L-1),2种改造结合得到化合物的Ki为1.4 nmol · L-1。旨在降低分子的亲脂性,选择氟原子取代苯环6位的氯原子得到化合物2(对TYK2的Ki= 1.6 nmol · L-1),能够改善TYK2的效力和选择性,维持代谢稳定。在小鼠炎症模型中,化合物2表现出对TYK2良好的抑制作用和阻断IL-12通路的潜能,以大于3 mg · kg-1的剂量经口给予化合物2可显著抑制IFN-γ。实验证明,在结构改造中不仅可以通过直接针对残基差异,还可以利用P-loop构象或灵活性的总体差异来获得药物选择性。
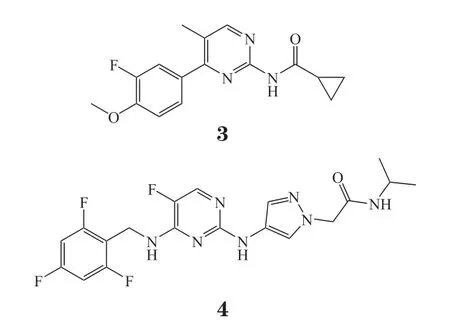
在2012年,Cellzome公司公开了关于嘧啶类似物作为TYK2抑制剂的专利[28],可用于治疗免疫性疾病、过敏和炎症。活性最优的化合物3能够高效抑制TYK2活性(IC50<100 nmol · L-1),且对TYK2选择性高于JAK2(IC50>10 μmol · L-1)。化合物4(IC50<100 nmol · L-1)嘧啶环的C4位被芳甲基氨基取代后对TYK2的选择性提高,对TYK2的活性超过对JAK1、JAK2和JAK3活性的100倍。
2016年,辉瑞公司公布了一系列含2,4-二氨基嘧啶环的TYK2抑制剂[29],其中嘧啶的4位上连有含氮多元环,2位氨基与六元芳环相连的化合物5对TYK2 的IC50为774 nmol · L-1。研究者通过优化4位含氮多元环和2位氨基上的芳基或杂芳基,可以调节TYK2活性和选择性,以五元芳杂环代替六元芳环,得到化合物6,其对TYK2 的IC50为23 nmol · L-1。

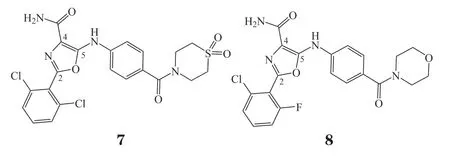
2015年,Sareum公司公开了有关5-氨基-2-苯基唑-4-甲酰胺结构的TYK2抑制剂专利[30],其用途涉及炎症和自身免疫性疾病。其中,化合物7抑制TYK2活性较好(对TYK2的IC50为1.9 nmol · L-1,对IL-23-pSTAT3的IC50为 51 nmol · L-1),超 过 其对JAK1、JAK2和JAK3酶的抑制活性(IC50分别为20、50和212 nmol · L-1)。化合物8能够有效抑制TYK2(IC50= 2.3 nmol · L-1),对TYK2的选择性高于对JAK1、JAK2和JAK3酶的活性(IC50分别为21.9、87.7和214 nmol · L-1)。最新的专利中,化合物8还可用于T细胞急性淋巴细胞性淋巴瘤的治疗[31]。
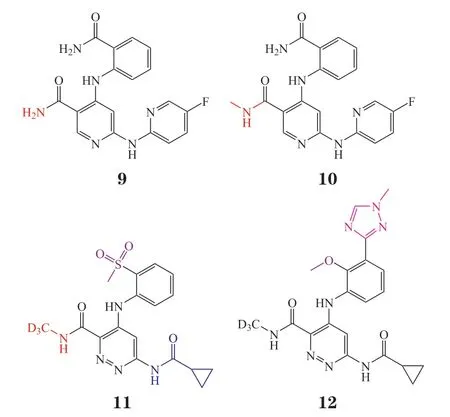
2018—2019年,百时美施贵宝(Bristol-Myers Squibb,BMS)公司公开了2项吡嗪酰胺结构的专利,保护选择性结合TYK2 JH2的变构抑制剂[32-33]。BMS通过高通量筛选得到具有烟酰胺结构的先导化合物9,其对TYK2 JH2的IC50为0.45 nmol · L-1,对TYK2/JAK1/JAK2 JH1的IC50分别为15、26和24 nmol · L-1,并采取了有效的药物化学策略,实现从先导化合物9,经化合物10和11到临床候选物12的转变[34]。化合物9引入甲基得到化合物10,其对TYK2 JH2的选择性有所提高,对TYK2 JH2 的IC50为1.3 nmol · L-1,对TYK2/JAK1/JAK2 JH1的IC50大于2 μmol · L-1。再通过引入氘增加代谢稳定性,用哒嗪环代替吡啶环增加通透性,用环丙酰胺取代减少人类Ether-a-go-go相关基因(human ether-a-gogo related gene,hERG)通道的抑制,以降低心血管反应,从而得到了化合物11,其对TYK2 JH2的IC50为1.3 nmol · L-1,对TYK2、JAK1、JAK2 JH1的IC50分别为大于50、大于2和大于50 μmol · L-1。在化合物11中引入甲基-1,2,4三氮唑和甲氧基,以提高代谢稳定性,并且与氨基酸形成重要氢键作用,增强与TYK2 JH2的亲和力,由此得到了化合物12。化合物12高选择性地与TYK2 JH2结合,其对TYK2 JH2的IC50为0.2 nmol · L-1,具有极佳的药物代谢动力学(pharmacokinetics,PK)性能,并在银屑病、结肠炎和红斑狼疮等自身免疫疾病模型中表现出极强的药效[35]。
化合物12作为新型口服选择性TYK2抑制剂,正进行关于斑块型银屑病的Ⅲ期临床试验。在已经完成的随机双盲、含安慰剂和活性对照(已上市银屑病治疗药物apremilast)的Ⅲ期临床试验POETYK PSO-1(NCT03624127)和POETYK PSO-2(NCT03611751)中,666例 和1 020例 中度至重度斑块型银屑病患者随机接受化合物12(6 mg,qd)、安慰剂或apremilast(30 mg,bid)的治疗。在治疗16周后, 服用化合物12的患者银屑病面积和严重程度指数改善75%(PASI75)比例分别为58.7%和53.6%,优于apremilast(35.1%和40.2%)以及安慰剂(12.7%和9.4%),在接受治疗24周后,这一比例提高到69%和59.3%。且有超过50%的患者皮肤症状完全或几乎完全消除。在安全性方面,化合物12的不良反应率也低于安慰剂和apremilast。这表明,化合物12有望成为银屑病患者新的治疗选择,通过口服给药是改善临床治疗的极具意义的举措[36]。BMS公司还在另外3项Ⅲ期研究(NCT04167462、NCT03924427和NCT04036435)中评估该药对其他疾病的疗效。关于系统性红斑狼疮、炎症性肠病等自身免疫性疾病的治疗应用也处于Ⅱ期临床研究中。
最近,研究人员报道了化合物13可作为与TYK2 JH2结合的TYK2抑制剂。通过对化合物13的甲基三氮唑基进行六元芳杂环取代的构效关系研究,发现对位氟原子的引入可以显著提高分子的活性(对TYK2 JH2 的IC50为0.19 nmol · L-1)和通透性,且对JAK家族的其他激酶具有高选择性。化合物13将在IL-23驱动的棘皮症、抗分化集群40(clusters of differentiation 40,CD40)诱导的结肠炎和自发性狼疮鼠模型中进行临床前研究[37]。

2.2 稠和杂环类
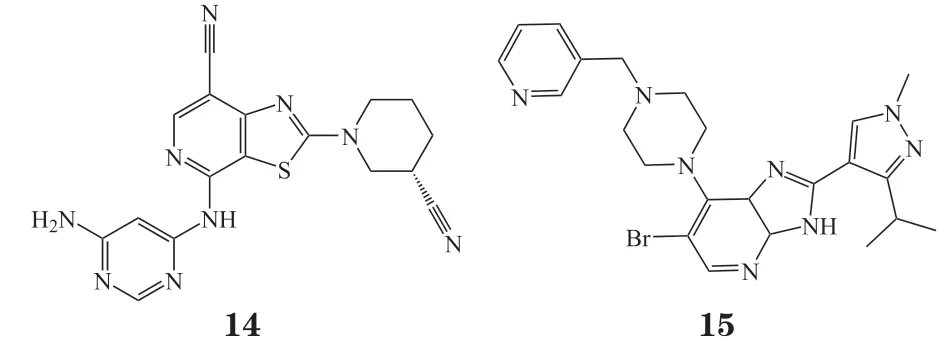
2015年基因泰克/罗氏公开了一项具有噻唑并[5,4-c]吡啶结构的TYK2抑制剂专利[38],代表性化合物14(Ki= 0. 2 nmol · L-1)有效抑制TYK2活性,可用于哮喘、炎症性肠病、类风湿性关节炎、银屑病的治疗。
同年,Jang等[39]通过基于片段的药物设计(fragment-based drug design,FBDD)发现了一种高效的TYK2抑制剂化合物15可以用来治疗类风湿关节炎。其完全抑制了由IFN-α介导的TYK2/STAT3信号传导途径以及下游磷酸化转录激活因子3(phosphorylation signal transducers and activators of transcription 3,pSTAT3)的表达,效果超过阳性化合物tofacitinib。
2017年,辉瑞公司基于TYK2 JH1 ATP位点的结构基序,开发了具有不同[5,6]-稠合杂环结构的化合物[24]。其中具有吡唑并[1,5-a]吡嗪环类化合物是有效的TYK2抑制剂[40]。化合物16在表现出一定TYK2选择性抑制活性的同时(TYK2 IC50= 10 nmol · L-1),具有较好的肝微粒体清除率(HLM) 为18 L · min-1· mg-1和亲脂性(LipE)为5.5。在此基础上,用环丁烷替代氮杂环丁烷得到了化合物17,共晶结构表明(见图3),顺式环丁腈部分与TYK2残基存在关键氢键和静电作用,能选择性抑制TYK2活性(IC50= 15 nmol · L-1),并大大降低肝微粒体清除率(HLM<8 L · min-1·mg-1)并提高通透性。在咪喹莫特诱发的皮炎小鼠模型中,化合物17能有效抑制IL-12分泌,灌胃给药分别为 1、3、10、 30 mg · kg-1(qd)时,IL-12 抑制率分别为51%、77%、 94%、98%。目前,化合物17作为选择性强效TYK2抑制剂,正在进行斑块型银屑病(NCT03895372)、溃疡性结肠炎(NCT04209556)和化脓性汗腺炎(NCT04092452)的临床研究。
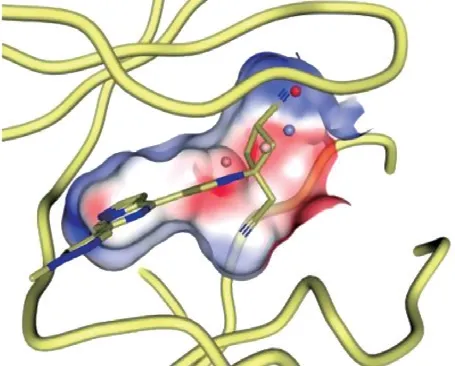
图 3 化合物17与TYK2共晶结构[24]Figure 3 Co-crystal structure of compound 17 and TYK2

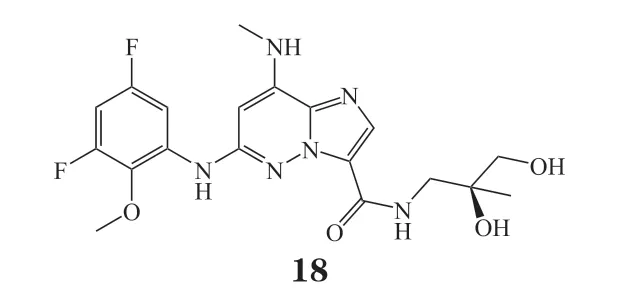
同年,BMS公司开发了具有咪唑[1,2-b]并哒嗪结构的化合物18(TYK2 JH2 IC50= 4 nmol · L-1),目前正在研究中,用于炎症和自身免疫性疾病的治疗[41]。
2018年,Nimbus Therapeutics公布了具有[5,6]-稠合多氮芳杂环结构的TYK2抑制剂专利[42],其中的活性最佳化合物19能有效抑制TYK2活性(TYK2 JH2 IC50<1 nmol · L-1),在雄性比格犬中以10 mg · kg-1经口给药,表现出良好的PK性质:血浆药物浓度-时间曲线下面积(AUC)为18 747 ng · mL-1· h,固有清除率(clearance,CLint)为0.62 mL · min-1· kg-1。目前Nimbus Therapeutics公司有2款TYK2抑制剂NDI-031301和NDI-031407 (结构未知)处于临床前研究中。TYK2的活化有利于T细胞急性淋巴细胞白血病(T-lymphocyte acute lymphoblastic leukemia,T-ALL)细胞的异常存活。NDI-031301是一种有效的选择性TYK2抑制剂,可抑制T-ALL细胞的增殖。给接种KOPT-K1 T-ALL细胞的免疫缺陷小鼠经口给药NDI-031301 100 mg · kg-1,可显著降低肿瘤负荷,提高生存效益[43]。NDI-031407在体外能够有效阻断TYK2介导的IL-23信号传导,并在小鼠模型中有效地抑制脊柱性关节炎(spondyloarthritis,SpA)的发展,因此其可作为潜在的治疗SpA的口服TYK2抑制剂[44]。
2019年,Abbvie公司宣布ABBV-712(结构未知)作为选择性TYK2抑制剂,开启银屑病Ⅰ期临床试验。在Abbvie公布的专利中[45],化合物20能有效抑制TYK2活性(EC50= 14 nmol · L-1),选择性高于JAK1(EC50= 3.4 μmol · L-1)和JAK2(EC50>25 μmol · L-1),适应证为银屑病和炎症性肠病。

同年,Sasaki等[46]报道了具有2,4-二氨基吡啶结构的新型大环类TYK2抑制剂。将化合物21吡咯烷酮环的4位与远端吡啶通过柔性饱和烷基键相连,可以保持化合物与TYK2的关键氢键作用(TYK2 IC50= 1.1 nmol · L-1、JAK1 IC50= 120 nmol · L-1、JAK2 IC50= 44 nmol · L-1、JAK3 IC50= 68 nmol · L-1)。其中大环化合物22对TYK2显示出强大的效力和极好的选择性(TYK2 IC50= 4.9 nmol · L-1、TYK2/JAK1活性比>140、TYK2/JAK2活性比>27、TYK2/JAK3 活性比>82),对IL-23也表现出较强的抑制活性(IC50= 210 nmol · L-1)。这些大环类TYK2抑制剂为选择性TYK2抑制剂的合成提供了新的设计思路(见图4)。

图 4 大环类TYK2抑制剂设计思路Figure 4 Ideas for the design of macrocyclic TYK2 inhibitors
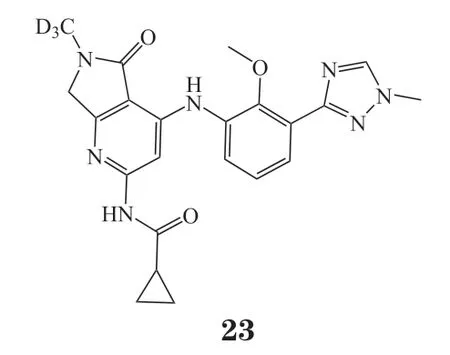
翰森医药近期也公布了一项选择性TYK2抑制剂专利,主要用途涉及皮炎、银屑病、类风湿关节炎、炎症性肠病、癌症等疾病[47]。代表化合物23结构与化合物13相似,差别在于吡啶环的C2与酰胺通过甲基相连形成[5,6]-稠合芳杂环结构。在人骨髓瘤细胞中,化合物23能有效抑制IFN-α TYK2-STAT3通路磷酸化信号传导(IC50= 1.25 nmol · L-1),对TYK2选择性高于JAK2调控的IL-6-pSTAT3途径(IC50= 88 nmol · L-1)。在雄性BALB/c小鼠经口给药试验中(5 mg · kg-1),化合物23表现出较好的PK性质[AUC = 3 654 ng · mL-1· h,半衰期(T1/2)= 2.5 h]。
3 结语与展望
近年来,TYK2作为JAK-STAT信号转导通路的关键激酶已在自身免疫疾病领域进行广泛的研究。选择性抑制TYK2,能够减少对机体其他免疫相关途径的抑制,降低感染风险,在临床上更加具有安全性。尽管暂时没有选择性TYK2抑制剂上市,但其能够在类风湿关节炎、强直脊柱炎、炎症性肠病、银屑病等自身免疫性疾病方面发挥独特作用,具有巨大的开发潜力。目前,小分子化合物12处于Ⅲ期临床试验阶段并且治疗效果显著、化合物17进入Ⅱ期临床试验阶段,有多个小分子在Ⅰ期临床及临床前生物活性测试阶段。相信不久之后,将会有选择性TYK2抑制剂成功上市,造福广大自身免疫疾病患者。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
湖北农业科学(2022年11期)2022-07-18
现代临床医学(2022年1期)2022-02-12
科学24小时(2018年1期)2018-01-10
中文信息(2017年2期)2017-04-13
江苏农业科学(2016年11期)2017-03-21
现代养生·下半月(2016年6期)2016-10-21
智能计算机与应用(2016年4期)2016-09-26
中国民族民间医药·下半月(2014年2期)2014-09-26
浙江中医杂志(2004年3期)2004-11-20
