
盘状镝簇合物的合成及缓慢磁弛豫
2022-07-12 07:40卫晓琴张凤英李万喜王新益张爱华刘毓芳
无机化学学报 2022年7期
卫晓琴 张凤英 李万喜 王新益 张爱华 刘毓芳
(1晋中学院材料科学与工程系,山西省轻质材料改性应用协同创新中心,晋中 030619)(2南京大学化学化工学院,配位化学国家重点实验室,南京 210023)(3晋中学院化学化工系,晋中 030619)
0 引 言
单分子磁体(single-molecule magnets,SMMs)是一类纳米尺度的磁性材料,在阻塞温度(TB)以下表现出缓慢的磁弛豫,不仅有类似磁体的磁滞行为,而且还具有丰富的量子磁学行为,如磁量子隧穿、磁量子相干和去相干等,在高密度信息存储[1]、量子计算[2]、分子开关以及分子自旋电子器件[3-4]等领域具有潜在的应用价值,因而备受研究者的关注。自Sessoli等[5]于1993年报道了首例超顺磁团簇化合物[Mn12]以来,SMMs登上了磁性研究的历史舞台,同时成为化学、物理乃至材料领域的研究焦点[6]。
为了获得性能优异的SMMs,即令其具有高的磁各向异性能垒(U)和TB,主要有2种方法:一种是增大化合物的基态自旋值S;另一种是增大化合物的单轴各向异性,使其具有较大的零场分裂参数(D)。早期研究者主要设计、合成具有强分子内相互作用的多核团簇金属化合物[7],期望通过增大化合物的S来提高U和TB。然而,大量的研究证实S的提高往往伴随着D的降低,从而达不到预期设计目标。直到2003年Ishikawa等报道了首例稀土基SMMs:[TbPc2]-[8],突破了SMMs的发展瓶颈,且得出一类新型的具有单个自旋中心的化合物,即单离子磁体(SIMs)。由于稀土离子的引入,相比于过渡金属簇,该化合物的TB显著提高。
磁各向异性是决定SMMs/SIMs缓慢磁弛豫的重要因素[9]。稀土离子对SMMs或SIMs弛豫机制会产生明显的贡献,有利于提高所得材料的磁各向异性,因此稀土单分子磁体从此登上了SMMs的历史舞台[10]。一方面,稀土离子的f电子由于其未猝灭的较大轨道角动量而具有相对较大的磁矩和磁各向异性,且同时存在旋轨耦合,因而相对于过渡金属离子而言,更有利于构筑高性能的SMMs。另一方面,由于稀土离子的f电子受外层s、d层电子的屏蔽,其顺磁离子之间磁相互作用较弱,从而易表现出SIMs的性质。此外,稀土离子中心的晶体场对SMMs的磁动力学行为至关重要[11]。晶体场的微弱改变能引起稀土金属离子基态能级分布的较大变化,进而影响SMMs/SIMs的磁弛豫行为。Long等分析了不同稀土离子4f电子的mj态电子云密度的形状,主要有扁圆形和扁长形[12]。其中TbⅢ和DyⅢ为典型的扁圆形基态电子云,在构建分子磁体时倾向于轴向配体场,从而确保其Stark亚能级基态具有大的轴各向异性,更容易获得高性能的SMMs。近年来,利用调控稀土离子配体场的策略,以单核和双核稀土SMMs为代表的稀土SMMs的研究取得了突破性的进展[13-16]。目前,SMMs的最大有效能垒(Ueff)达到2 219 K,其最高阻塞温度为80 K[17],已突破了液氮的沸点温度(77 K),这为开发和研究室温下的SMMs奠定了坚实的基础,推动了分子磁性材料在信息领域的实际应用进程。
稀土基SMMs的研究主要集中在4f体系[18]、4f-4f体系[19]、3d-4f体系[20]和 2p-3d-4f体系[21],目前还未涉及4d-4f体系。在4d金属中,MoⅢ离子具有低自旋的d3电子组态,S为1/2,与氰根形成[MoⅢ(CN)7]4-构筑块。MoⅢ的单离子各向异性及各向异性磁耦合使得[MoⅢ(CN)7]4-单元具有很强的磁各向异性,使其在构筑高阻塞温度的SMMs/SIMs方面非常有前途[22]。在钼基分子磁性材料中,主要研究成果为3d-4d体系。利用3d和4d金属离子之间强的各向异性磁耦合及[MoⅢ(CN)7]4-构筑块提供的D5h配体场,获得了一系列突出的研究成果[23-27]。
我们用稀土离子、双齿配体(图1)和[MoⅢ(CN)7]4-构筑块进行自组装,获得了一例MoⅢ被氧化成[MoⅥ(L)O(CN)3]+的分子团簇,其分子式为[DyⅢ7(tmphen)12O6(OH)6Cl2][MoⅥ(tmphen)O(CN)3]6Cl7·66H2O(1,tmphen=3,4,7,8-四甲基-1,10-菲咯啉)。对该化合物进行了晶体结构和磁性研究,结果表明化合物1形成了七核镝的团簇结构,类似圆盘状,且表现出SMMs的性质。

图1 配体tmphen结构示意图Fig.1 Schematic view of ligand tmphen
1 实验部分
1.1 试剂及主要仪器
所用初始原料K4Mo(CN)7·2H2O参考文献合成[28],其他试剂如稀土盐和配体tmphen以及所用溶剂均为市售分析纯,未经进一步纯化。
由于K4Mo(CN)7·2H2O容易被氧化,所有实验操作均在充满高纯氮气的手套箱中完成。元素分析使用Vario MICRO型元素分析仪进行。红外光谱使用德国Bruker Tensor 27 FT-IR傅里叶变换红外光谱仪(固体 KBr压片法,400~4 000 cm-1范围)测试。单晶衍射数据使用带有石墨单色器的德国Bruker APEX Duo CCD衍射仪测定。磁性数据使用美国Quantum Design公司SQUID VSM超导量子干涉磁性测量系统测定。粉末X射线衍射(PXRD)数据使用德国Bruker D8 Advance X射线粉末衍射仪(CuKα射线,λ=0.154 18 nm,40 kV,40 mA,2θ=5°~50°)测定。热重分析(TGA)使用德国Netzsch STA 449 C热重-差热同步分析仪(升温速率为10℃·min-1,温度范围在40~800℃)进行。
1.2 化合物1的合成
将 K4Mo(CN)7·2H2O(0.04 mmol,18.8 mg)溶解于9 mL 乙腈和水的混合溶剂(VMeCN∶VH2O=1∶1)中,DyCl3·6H2O(0.1 mmol,约 38 mg)和 tmphen(0.2 mmol,47 mg)溶解于9 mL相同比例的混合溶剂中,两者混合得到红棕色溶液。过滤掉不溶物,滤液置于玻璃瓶中,密封静置。一个月后,玻璃瓶底部有红棕色块状晶体生成,并有一些棕色粉末沉淀。过滤收集晶体,用母液反复洗涤至干净,干燥得15 mg左右,产率约为 27%(根据 Mo3+计算)。样品(Dy7Mo6C306H426Cl9N54O84)的元素分析测试实验值(理论值 ,%):C,46.57(44.62);H,4.88(5.21);N,9.73(9.18)。红外特征吸收峰(KBr,cm-1):3 421(s),2 922(m),2 865(m),2 482(s),2 095(s),1 619(s),1 591(m),1 527(vs),1 428(vs),1 386(s),1 307(w),1 272(w),1 244(m),1 177(vw),1 089(vw),1 016(w),965(s),863(m),818(m),726(vs),620(w),524(w),461(w),446(vw)。
1.3 X射线单晶衍射分析
单晶衍射数据在带有石墨单色器的德国Bruker APEX Duo CCD衍射仪上测定,CCD衍射仪采用MoKα射线(λ=0.071 073 nm)、ω扫描方式进行数据收集。使用惰性油Paratone包裹晶体进行测试,并使用APEX Ⅱ程序确定晶胞参数和收集数据。所得数据用CrysAlisPro软件进行数据还原和吸收校正处理[29]。晶体数据使用Olex2[30]软件、采用SHELXT程序进行直接法解析,并在基于F2全矩阵最小二乘法的基础上,使用SHELXL-2018程序进行精修[31]。所有非氢原子都进行了各向异性热参数精修。有机配体的氢原子是通过理论加氢获得。晶体结构空洞中存在氯离子和大量溶剂水分子,使用SQUEEZE进行处理。晶体结构收集和精修参数见表1,部分键长、键角见表2。

表1 化合物1的晶体结构数据和精修参数Table 1 Crystal data and structure refinement for compound 1

表2 化合物1的部分键长(nm)和键角(°)Table 2 Selected bond distances(nm)and bond angles(°)for compound 1
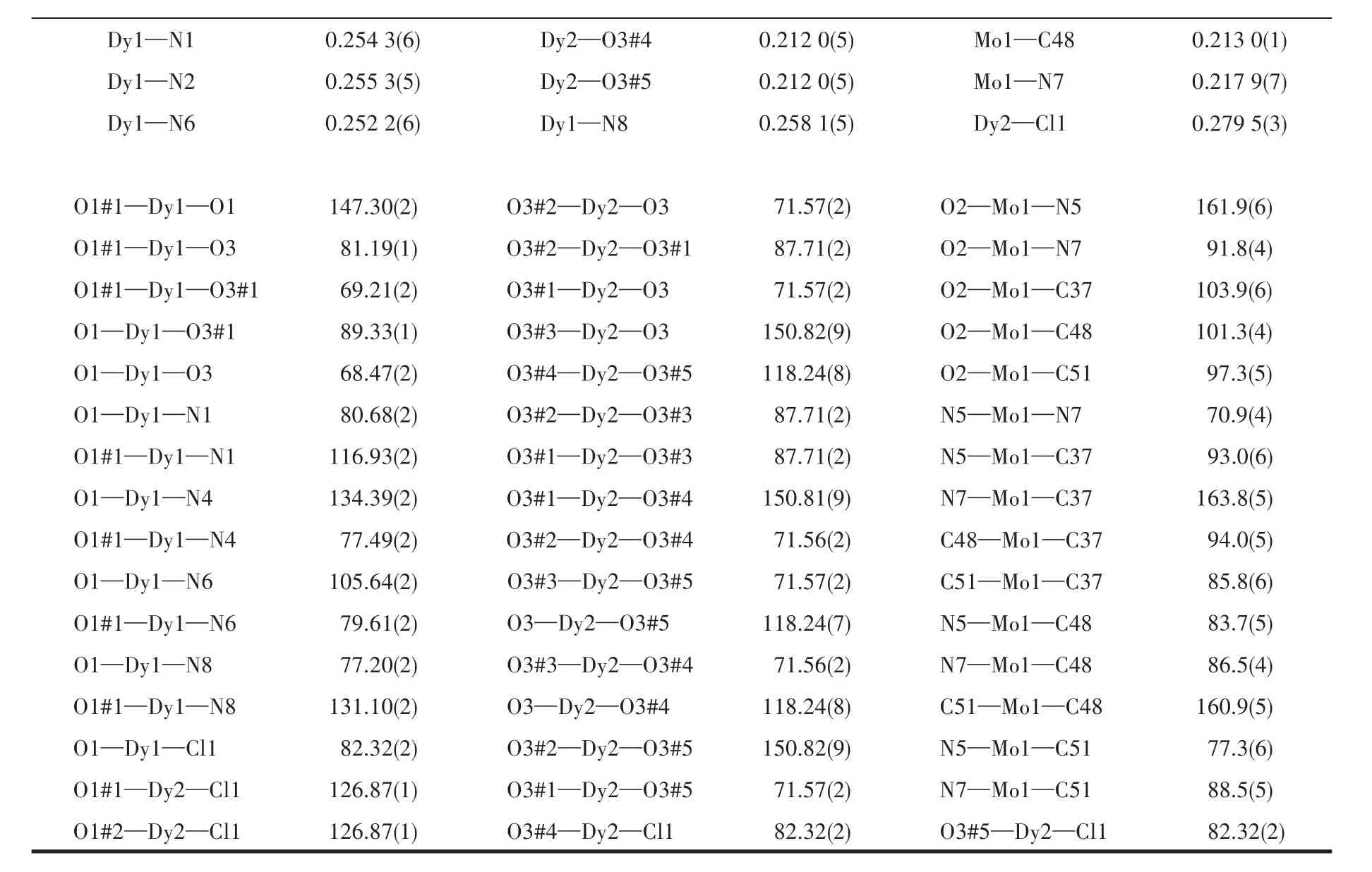
续表2
CCDC:2161872。
2 结果与讨论
2.1 晶体结构
化合物1的晶体结构属于三方R3空间群,其分子结构如图2所示。该化合物的主体部分是一个含中心的六边形团簇,类似于圆盘状结构,如图3所示。7个Dy3+离子具有2种晶体学位置:其中6个Dy3+离子(Dy1~Dy6)形成外围的六边形,它们几乎在同一个平面上;1个Dy3+离子(Dy7)位于该六边形的中心位置,但是它与Dy6环不在同一个平面上,而是无序成2个位点分布在Dy6环平面的两侧位置。6个外围的Dy3+离子之间通过6个(μ2-O2-)基团彼此连接,形成了一个近似规则的六边形(所有的Dy—Dy—Dy键角接近 120°,Dy—Dy距离近似为 0.37 nm)。外围的Dy3+离子和中心Dy3+离子通过6个μ3-OH-基团连接,这些基团一上一下交替分布在Dy7平面外。6个外围的稀土离子Dy1~Dy6分别和2个tmphen配体配位,形成了[N4O4]的八配位环境,配位构型为畸变的三角十二面体。Dy—O的键长范围为0.227 2(4)~0.241 9(4)nm,Dy—N 的键 长 范围为0.252 2(6)~0.258 1(5)nm。这 6 个 Dy3+离子是对称的,它们的CShMs(continuous shape measures)偏离值[32]均为1.463。中心的Dy7离子处于[O6Cl]的七配位环境,配位构型为畸变的三角四面体,其CShMs偏离值为5.691;Dy—O的键长范围为0.212 0(5)~0.262 7(6)nm,Dy—Cl键长为0.279 5(3)nm。

图2 化合物1的晶体结构Fig.2 Crystal structure of compound 1

图3 化合物1在ab平面的圆盘状结构(左)和空间填充图(右)Fig.3 Disc-like structure of 1 in the ab plane(left)and spatial packing diagram(right)
尽管实验操作是在氮气氛围的手套箱中进行,但是极微量的氧气仍然可以使MoⅢ发生氧化。根据Mo的配位键进行的价键计算(BVS)[33-35]表明Mo的氧化数为+6,形成了[MoⅥ(tmphen)O(CN)3]+阳离子游离在晶格中。该化合物的结构中包含6个[MoⅥ(tmphen)O(CN)3]+阳离子,SQUEEZE处理的部分包含7个Cl-离子和66个H2O分子。此外,由于化合物1使用的tmphen是芳香环配体,形成的盘状团簇中具有弱的π-π相互作用,芳香环中心的距离在0.356 5~0.371 9 nm之间,如图4所示。
2.2 热稳定性和纯度
为了确定化合物1的稳定性,我们采用热重分析仪研究了其在氮气气氛下的热分解行为。如图5所示,化合物1的TGA曲线显示该化合物在0~200℃损失约11.77%的重量,对应着晶胞中66个水分子的失去(理论值为14.42%)。300℃以后,随着温度的升高,晶体开始分解。TGA结果进一步证实了晶体结构中水分子的个数。

图5 化合物1的TGA曲线Fig.5 TGA curve of compound 1
为了验证化合物1的纯度,我们在室温下对该化合物进行了PXRD表征,结果如图6所示。实验测试结果与单晶模拟结果的出峰位置基本吻合,说明得到的化合物为纯相。
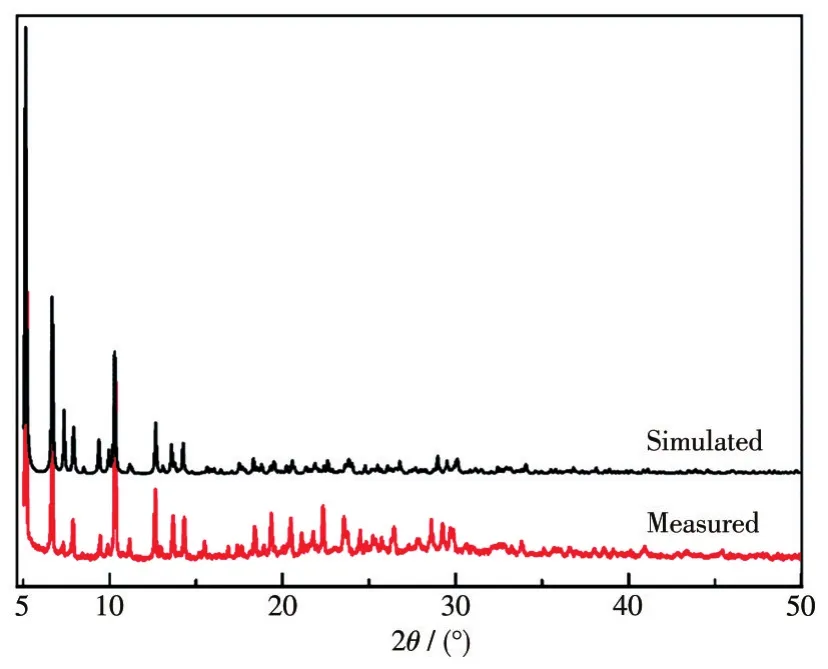
图6 化合物1的PXRD图Fig.6 PXRD patterns of compound 1
2.3 磁学性质
在1 kOe直流场下、2~300 K温度范围测试了化合物1的变温直流磁化率曲线,结果如图7所示。室温χMT值为91.63 cm3·mol-1·K,接近于Dy7单元的净 自旋值 99.19 cm3·mol-1·K(χMT=7[gJ2J(J+1)]/8,gJ=15/2,J=4/3,g=2.0)。随着温度的降低,χMT曲线先缓慢减小,至20 K后突然减小。该现象表明Dy3+离子之间可能存在弱的反铁磁相互作用,或者是由Dy3+离子的热致激发态Stark子能级去布居导致[36]。化合物1在不同温度下的磁化强度曲线见图7和图8a。低温下M-H曲线在70 kOe时近似达到饱和值37μB,随着温度的升高,70 kOe时的磁化强度值逐渐减小,这种现象表明该化合物具有强的磁各向异性或者存在低能量的激发态。然而,该化合物在低温下没有明显的磁滞回线(图8b)。

图7 化合物1的变温直流磁化率曲线和不同温度下的磁化强度曲线(插图)Fig.7 Temperature-dependent direct-current susceptibility curve and magnetization curves at different temperatures(Inset)of compound 1

图8 化合物1的M-HT-1曲线(a)和磁滞回线(b)Fig.8 M-HT-1curves(a)and magnetic hysteresis loop(b)of compound 1
为了探索化合物1的磁动力学,我们在零场下测试了它的变温交流磁化率和变频交流磁化率,结果如图9所示。交流磁化率的实部χM′和虚部χM″信号均表现出明显的频率依赖和温度依赖,说明该化合物具有缓慢的磁弛豫行为,是典型的SMMs。根据变频交流磁化率数据,获得了该化合物从4.0~20.0 K的Cole-Cole曲线,如图10a所示。此外,通过广义的Debye模型对Cole-Cole曲线进行拟合,可获得不同温度下的弛豫时间τ和弛豫的分布参数α(表3)。从表3可以看出,α值在0.16~0.42之间,说明弛豫时间具有一定的分布。根据Arrhenius公式:τ=τ0exp[Ueff/(kT)],可以得到 lnτ对T-1的 Arrhenius曲线(图10b)。对表3中高于6.0 K的数据进行线性拟合,获得了该化合物的Ueff为 51.6 K(35.8 cm-1),τ0为 17 μs。从图10b可以看出,当温度低于6.0 K时,lnτ偏离线性分布并趋于饱和,表明该体系中可能存在量子隧穿弛豫路径,也正好解释了化合物1在低温下没有磁滞回线的现象。

图9 化合物1的变温交流磁化率曲线(a)和变频交流磁化率曲线(b)Fig.9 Temperature-dependent(a)and frequency-dependent(b)alternating current susceptibility curves of compound 1

图10 (a)化合物1的Cole-Cole曲线,实线表示根据Debye模型拟合的结果;(b)ln τ vs T-1图,红色实线代表Arrhenius线性拟合Fig.10 (a)Cole-Cole curves of 1,where solid lines represent the fitting results according to the Debye model;(b)ln τ vs T-1plot,where solid red line represents the Arrhenius linear fitting

表3 根据广义德拜模型拟合Cole-Cole曲线的参数Table 3 Fitting parameters for Cole-Cole curves according to the generalized Debye model*
3 结 论
采用原位合成法将DyCl3·6H2O、配体tmphen以及[MoⅢ(CN)7]4-构筑块组装获得一例盘状七核镝团簇化合物。由于MoⅢ对空气敏感,自组装过程中[MoⅢ(CN)7]4-构筑块发生氧化分解形成了[MoⅥ(tmphen)O(CN)3]+模块,充当抗衡阳离子游离在晶格中。芳香环配体的使用使得形成的盘状团簇化合物中具有弱的π-π相互作用。磁性测量表明该化合物中存在量子隧穿弛豫路径,使其低温下没有出现磁滞回线的现象。零场下的交流磁化率数据表明该化合物具有单分子磁体的性质,有效能垒达到 51.6 K(35.8 cm-1,τ0=17 μs)。在后续实验中,期望将[MoⅢ(CN)7]4-构筑块成功组装到多核团簇化合物中,获得4d-4f型单分子磁体。
猜你喜欢
天津农学院学报(2022年2期)2022-08-05
渤海大学学报(自然科学版)(2022年1期)2022-07-25
云南大学学报(自然科学版)(2022年3期)2022-05-25
表面工程与再制造(2021年1期)2021-08-06
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
科学大观园(2019年13期)2019-09-10
科学中国人(2017年36期)2017-06-09
科技创新导报(2016年30期)2017-03-15
中学生数理化·高三版(2016年2期)2016-09-10
